Executive Summary
P21 (also designated P021 or Peptide 021) is a synthetic tetrapeptide derived from the biologically active region of ciliary neurotrophic factor (CNTF) that has demonstrated consistent neurogenic and neurotrophic effects across multiple preclinical models of neurodegenerative disease, cognitive aging, and developmental disorders.
Key Takeaways
- Derived from CNTF residues 148-151 with adamantane modification for BBB penetration
- Upregulates BDNF expression through LIF signaling inhibition
- Reduces tau hyperphosphorylation via GSK-3 beta inhibition
- Rescued cognitive deficits in 3xTg-AD mice even after pathology onset
- 18-month chronic treatment in mice showed no adverse effects
The peptide was developed in the laboratory of Dr. Khalid Iqbal at the New York State Institute for Basic Research in Developmental Disabilities. Its design involved epitope mapping of CNTF to identify the minimal active sequence (amino acid residues 148-151), followed by addition of an adamantylated glycine at the C-terminus to improve blood-brain barrier permeability and resistance to enzymatic degradation. The resulting compound, Ac-DGGLAG-NH2, has a molecular weight of 578.3 daltons and retains the neurotrophic properties of full-length CNTF without the serious side effects that derailed clinical development of the parent molecule.
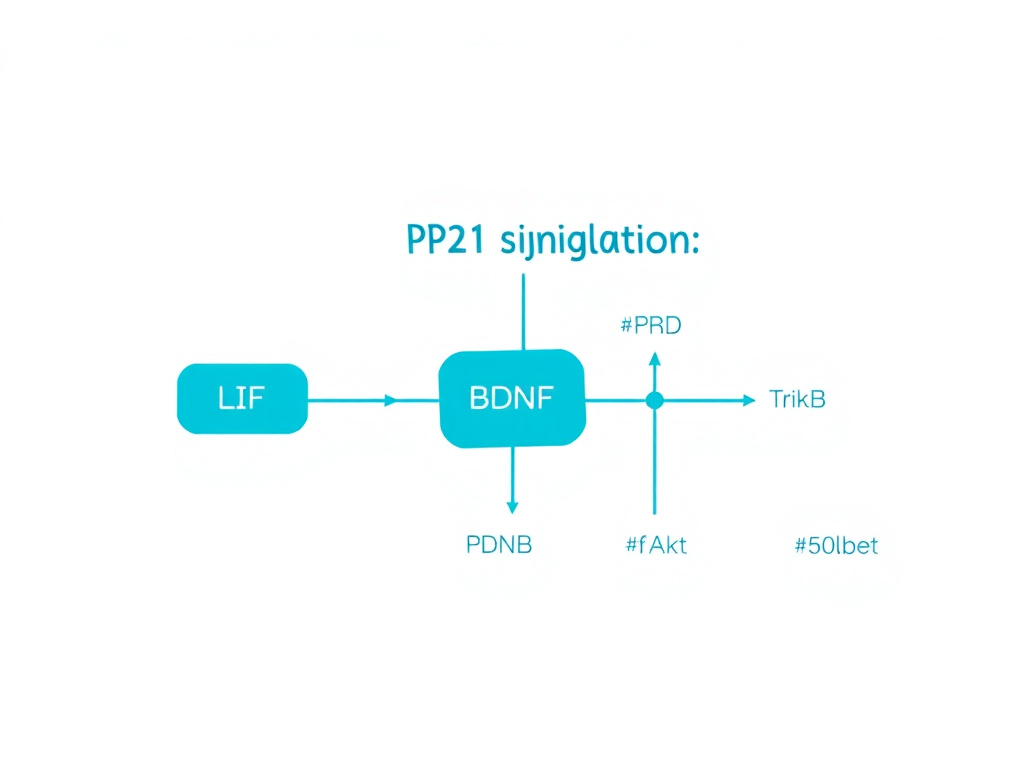
What makes P21 particularly interesting among nootropic peptides is its dual mechanism of action. It simultaneously promotes the birth of new neurons (neurogenesis) in the hippocampal dentate gyrus while also protecting existing neurons from degeneration. This is accomplished through inhibition of leukemia inhibitory factor (LIF) signaling, which in turn upregulates brain-derived neurotrophic factor (BDNF) expression. The downstream cascade activates the TrkB/PI3K/Akt pathway, leading to inhibitory phosphorylation of glycogen synthase kinase-3 beta (GSK-3 beta) at serine 9, which prevents tau hyperphosphorylation, one of the hallmark pathological features of Alzheimer's disease.
In the triple-transgenic Alzheimer's disease mouse model (3xTg-AD), chronic P021 treatment rescued cognitive impairment, restored synaptic plasticity, reduced both amyloid-beta and hyperphosphorylated tau pathology, and boosted hippocampal neurogenesis. These effects were observed even when treatment began after amyloid pathology had already developed, suggesting disease-modifying rather than merely preventive potential. Long-term treatment spanning up to 18 months in mice produced no weight loss, tumors, or signs of pain, a sharp contrast to the anorexia, muscle cramps, and weight loss caused by full-length CNTF.
The compound is orally bioavailable with over 90% stability in gastric juice and greater than 97% stability in intestinal fluid at body temperature. Its plasma half-life exceeds three hours in mice. Despite these promising pharmacokinetic properties, P021 has not yet entered human clinical trials. Phanes Biotech, co-founded by Dr. Iqbal, is currently advancing the compound toward clinical development for Alzheimer's disease and related conditions.
Key Research Highlights
- Derived from CNTF residues 148-151 with adamantane modification for BBB penetration
- Upregulates BDNF expression through LIF signaling inhibition
- Reduces tau hyperphosphorylation via GSK-3 beta inhibition
- Rescued cognitive deficits in 3xTg-AD mice even after pathology onset
- 18-month chronic treatment in mice showed no adverse effects
- Orally bioavailable with plasma half-life over 3 hours
- No human clinical trials have been conducted to date
This report examines the complete body of published research on P21, covering its molecular origins, mechanisms of action, preclinical efficacy data across multiple disease models, safety profile, dosing considerations, and how it compares to related nootropic compounds like Semax, Dihexa, and the parent preparation Cerebrolysin. Every claim is tied to published peer-reviewed research, and the limitations of the current evidence base, which remains entirely preclinical, are clearly noted throughout.
CNTF-Derived Design: From Neurotrophic Factor to Druggable Peptide
The Problem with Full-Length Neurotrophic Factors
Neurotrophic factors, including CNTF, brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), and glial cell line-derived neurotrophic factor (GDNF), have long been recognized as promising therapeutic candidates for neurodegenerative diseases. Their ability to promote neuronal survival, stimulate neurite outgrowth, and enhance synaptic plasticity makes them attractive targets. However, their clinical application has been severely limited by several pharmacological challenges that have proven difficult to overcome.
First, these large protein molecules cannot cross the blood-brain barrier (BBB) when administered peripherally. This necessitates invasive delivery methods such as intracerebroventricular infusion, which carries infection risk and requires surgical implantation of delivery devices. Second, full-length neurotrophic factors have short half-lives in vivo and are rapidly degraded by proteases. Third, and perhaps most critically, systemic administration of these proteins produces serious adverse effects.
CNTF provides a particularly instructive case study. Clinical trials of recombinant CNTF for amyotrophic lateral sclerosis (ALS) in the 1990s were plagued by dose-limiting toxicities. Patients experienced significant anorexia, severe muscle cramps, hyperalgesia (increased pain sensitivity), and substantial weight loss. These side effects are attributed to CNTF's ability to activate an alternative receptor complex involving IL-6R alpha, LIFR beta, and gp130, which triggers inflammatory and catabolic signaling pathways independent of its neurotrophic effects. The disconnect between CNTF's beneficial neurotrophic actions and its harmful systemic effects motivated the search for smaller, more selective molecules that could retain the former while eliminating the latter.
Epitope Mapping and Minimal Active Sequence Identification
The development of P021 began with a systematic effort to identify the minimal region of CNTF responsible for its neurotrophic activity. Dr. Khalid Iqbal and colleagues at the New York State Institute for Basic Research employed epitope mapping, a technique that involves synthesizing overlapping peptide fragments covering the entire CNTF sequence and testing each fragment for biological activity.
CNTF is a 200-amino-acid protein that belongs to the interleukin-6 (IL-6) family of cytokines. It signals primarily through a tripartite receptor complex consisting of CNTF receptor alpha (CNTFR alpha), LIFR beta, and gp130. The neurotrophic effects are mediated through downstream activation of the JAK-STAT signaling pathway, particularly STAT3 phosphorylation. Through their epitope mapping work, the researchers identified that amino acid residues 148-151 of human CNTF constituted the core bioactive region responsible for the protein's neurotrophic and neurogenic properties.
This was a significant finding because it meant that a tetrapeptide, just four amino acids long, could potentially replicate the beneficial effects of the entire 200-amino-acid protein. However, a bare tetrapeptide would face two major obstacles in vivo: rapid degradation by exopeptidases and poor blood-brain barrier penetration due to its hydrophilic character.

The Adamantane Solution
To solve both problems simultaneously, the researchers attached an adamantylated glycine residue to the C-terminus of the tetrapeptide. Adamantane is a diamondoid hydrocarbon with a cage-like molecular structure that confers several pharmacologically advantageous properties. Its lipophilic character dramatically increases the peptide's ability to cross cellular membranes, including the blood-brain barrier. At the same time, the bulky adamantane group sterically hinders exopeptidases from cleaving the peptide's terminal amino acids, substantially increasing its metabolic stability.
The resulting molecule, designated P021 (Ac-DGGLAG-NH2), has a molecular weight of 578.3 daltons and combines the biological activity of CNTF's core epitope with drug-like pharmacokinetic properties. The N-terminal acetylation (Ac-) provides additional protection against aminopeptidases, while the C-terminal amidation (-NH2) further enhances stability and receptor binding.
Pharmacokinetic studies in mice confirmed that the design achieved its goals. P021 demonstrated over 90% stability in artificial gastric juice after 30 minutes, greater than 97% stability in artificial intestinal fluid after 2 hours at 37 degrees Celsius, and a plasma half-life exceeding 3 hours. The compound successfully penetrated the blood-brain barrier following oral administration, as confirmed by detection of the intact peptide in brain tissue.
Relationship to Cerebrolysin
It's worth clarifying the relationship between P021 and Cerebrolysin. While P021 is sometimes described as "cerebrolysin-derived," the relationship is more nuanced. Cerebrolysin is a complex mixture of low-molecular-weight neuropeptides and free amino acids derived from enzymatic hydrolysis of porcine brain proteins. It contains fragments of multiple neurotrophic factors, including CNTF, BDNF, NGF, and GDNF. The neurotrophic activity of Cerebrolysin is attributed in part to these CNTF-derived fragments, but the preparation contains hundreds of distinct peptides.
P021 represents a rational, reductionist approach to capturing the essential neurotrophic activity present in Cerebrolysin. Rather than using the entire complex mixture, the researchers isolated the specific CNTF epitope responsible for neurogenic activity and optimized it for drug-like properties. In this sense, P021 can be understood as a next-generation compound that distills the most relevant mechanism of Cerebrolysin into a single, well-characterized molecule. This approach offers advantages in terms of reproducibility, quality control, regulatory pathway, and mechanistic understanding, all of which are challenging with a complex biological preparation like Cerebrolysin.
CNTF vs. P021: Design Rationale
Full-length CNTF (200 amino acids) cannot cross the BBB, has a short half-life, and causes anorexia and weight loss through activation of inflammatory signaling cascades. P021 (6 amino acids including the adamantylated glycine) retains the core neurotrophic activity while crossing the BBB, resisting proteolytic degradation, and avoiding the systemic toxicity of the parent molecule. In 18 months of chronic treatment in mice, P021 produced no weight loss, tumors, or signs of pain.
Molecular Structure and Chemical Properties
| Property | Value |
|---|---|
| Full Name | Peptide 021 (P021) |
| Sequence | Ac-DGGLAG-NH2 (Acetyl-Asp-Gly-Gly-Leu-Ala-Gly(adamantane)-NH2) |
| Molecular Weight | 578.3 Da |
| Parent Molecule | Human CNTF (residues 148-151) |
| N-terminal Modification | Acetylation (protection from aminopeptidases) |
| C-terminal Modification | Adamantylated glycine + amidation |
| Gastric Stability (30 min, 37C) | > 90% |
| Intestinal Stability (2 hr, 37C) | > 97% |
| Plasma Half-life (mice) | > 3 hours |
| BBB Penetration | Confirmed (oral and peripheral routes) |
| Oral Bioavailability | Yes (demonstrated in rodent models) |
Neurogenesis Mechanism: How P21 Drives New Neuron Formation
P21's ability to stimulate neurogenesis in the adult hippocampus is its most distinctive pharmacological property and the feature that most clearly differentiates it from other nootropic peptides. Understanding this mechanism requires examining a cascade of molecular events that begins with LIF inhibition and culminates in the birth, survival, and functional integration of new neurons in the dentate gyrus.
The LIF-BDNF Axis: P21's Primary Target
Leukemia inhibitory factor (LIF) is a pleiotropic cytokine that belongs to the same IL-6 family as CNTF. In the adult brain, LIF signaling through the JAK-STAT3 pathway exerts a tonic inhibitory effect on neurogenesis. Under normal physiological conditions, LIF helps maintain the balance between neural stem cell quiescence and proliferation. However, in aging and neurodegenerative disease, elevated LIF signaling contributes to suppressed neurogenesis, reduced BDNF expression, and impaired synaptic plasticity.
P021 acts as a competitive inhibitor of LIF signaling through the JAK-STAT3 pathway. By blocking LIF's interaction with its receptor complex, P021 releases the brake on neurogenesis and simultaneously removes the suppressive influence on BDNF transcription. The result is a significant increase in BDNF mRNA and protein levels in the hippocampus, particularly in the dentate gyrus where adult neurogenesis occurs.
This increase in BDNF is not merely incidental to P21's effects. It represents the central mechanism through which the peptide exerts its neurogenic, neuroprotective, and cognitive-enhancing actions. BDNF is the most abundant neurotrophin in the adult brain and plays a critical role in neuronal survival, dendritic branching, spine formation, long-term potentiation (LTP), and memory consolidation. Age-related decline in BDNF levels has been consistently associated with cognitive impairment, hippocampal atrophy, and increased vulnerability to neurodegenerative disease.
The BDNF/TrkB/PI3K/Akt/GSK-3 beta Signaling Cascade
Once BDNF levels are elevated by P21-mediated LIF inhibition, the neurotrophin binds to its high-affinity receptor, tropomyosin receptor kinase B (TrkB). This triggers receptor dimerization and autophosphorylation, initiating several downstream signaling cascades. The pathway most relevant to P21's therapeutic effects is the PI3K/Akt pathway.
Activated TrkB recruits and activates phosphoinositide 3-kinase (PI3K), which generates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) at the cell membrane. PIP3 recruits protein kinase B (Akt) to the membrane, where it is phosphorylated and activated by phosphoinositide-dependent kinase 1 (PDK1). Active Akt then phosphorylates glycogen synthase kinase-3 beta (GSK-3 beta) at serine 9, which is an inhibitory phosphorylation that reduces GSK-3 beta kinase activity.
This inhibition of GSK-3 beta is therapeutically significant for two reasons. First, GSK-3 beta is one of the primary kinases responsible for tau hyperphosphorylation, the process that leads to neurofibrillary tangle formation in Alzheimer's disease. By inhibiting GSK-3 beta, P21 directly reduces the pathological phosphorylation of tau protein. Second, GSK-3 beta activity suppresses adult neurogenesis through multiple mechanisms, including inhibition of Wnt/beta-catenin signaling and promotion of neural progenitor cell apoptosis. Reducing GSK-3 beta activity therefore creates a more permissive environment for neurogenesis.
Dentate Gyrus Neurogenesis: Evidence from Animal Studies
The hippocampal dentate gyrus is one of only two regions in the adult mammalian brain where neurogenesis continues throughout life (the other being the subventricular zone lining the lateral ventricles). New neurons are born from neural stem cells in the subgranular zone (SGZ) of the dentate gyrus, migrate a short distance into the granule cell layer (GCL), and over a period of several weeks mature into functionally integrated granule neurons that participate in hippocampal circuits involved in pattern separation, spatial memory, and contextual learning.
Multiple studies have demonstrated that P021 treatment significantly increases neurogenesis in the dentate gyrus. The evidence comes from several complementary markers:
- Ki-67: A marker of cell proliferation expressed during active phases of the cell cycle. P021 treatment significantly increased the number of Ki-67-positive cells in the SGZ and GCL of both wild-type and 3xTg-AD mice, indicating enhanced proliferation of neural progenitor cells.
- Doublecortin (DCX): A microtubule-associated protein expressed by immature neurons during the first 2-3 weeks of neuronal differentiation. P021 increased DCX-positive cell counts, confirming that the newly proliferating cells were committing to a neuronal fate rather than becoming glia.
- BrdU (Bromodeoxyuridine) incorporation: BrdU is a thymidine analog that gets incorporated into DNA during S-phase of cell division. Animals treated with P021 showed significantly higher numbers of BrdU-positive cells in the dentate gyrus, and co-labeling with the mature neuronal marker NeuN confirmed that these cells had differentiated into functional neurons.
- NeuN co-labeling: The co-expression of BrdU and NeuN in the same cells demonstrates that newly born cells (BrdU-positive) have matured into neurons (NeuN-positive), establishing that P021 not only increases cell proliferation but promotes neuronal maturation and survival.
In the 3xTg-AD mouse model, where neurogenesis is significantly impaired compared to wild-type animals, P021 treatment restored neurogenesis to near-normal levels. This rescue of neurogenic capacity was observed even when treatment began at 9-10 months of age, a time point when amyloid pathology is already established in these mice. The neurogenic effects persisted through at least 18 months of continuous treatment, with assessments at 15-16 months and 21-22 months of age showing sustained increases in proliferation and neuronal differentiation markers.
Synaptic Plasticity and Dendritic Remodeling
Beyond promoting the birth of new neurons, P021 also enhances the structural and functional plasticity of existing neurons. This is consistent with the compound's effect on BDNF, which is a master regulator of synaptic plasticity. Several lines of evidence support this:
Dendritic complexity: Golgi staining analysis in P021-treated 3xTg-AD mice revealed rescue of dendritic deficits in CA1 pyramidal neurons. Treated animals showed increased dendritic branching and total dendritic length compared to vehicle-treated controls, approaching values seen in wild-type animals.
Spine density: Dendritic spine density, a morphological correlate of excitatory synapse number, was significantly increased by P021 treatment. This is consistent with BDNF's well-established role in promoting spinogenesis and spine maintenance.
Synaptic protein expression: P021 treatment increased expression of pre-synaptic markers (synaptophysin, synapsin I) and post-synaptic markers (PSD-95) in the hippocampus of 3xTg-AD mice. These proteins are essential for normal synaptic transmission and are typically reduced in Alzheimer's disease.
CREB phosphorylation: Cyclic AMP response element-binding protein (CREB) is a transcription factor that regulates genes involved in synaptic plasticity and memory consolidation. P021 treatment increased phosphorylated CREB (p-CREB) levels in the hippocampus, indicating enhanced transcriptional activation of plasticity-related genes.
Clinical Relevance of P21's Dual Mechanism
P21's simultaneous promotion of neurogenesis and synaptic plasticity through BDNF upregulation addresses two of the major deficits in Alzheimer's disease and age-related cognitive decline. Most current Alzheimer's drugs (cholinesterase inhibitors, memantine) provide only symptomatic relief without addressing underlying pathology. P21's ability to reduce tau and amyloid pathology while also promoting structural brain repair represents a fundamentally different therapeutic approach. However, all evidence to date comes from animal models, and translation to human efficacy remains unproven.
The CREB-BDNF Positive Feedback Loop
An additional feature of P21's mechanism that deserves attention is the existence of a positive feedback loop between CREB activation and BDNF expression. BDNF gene transcription is regulated by CREB, meaning that P021-mediated increases in CREB phosphorylation further enhance BDNF production. This creates a self-amplifying cycle: P021 inhibits LIF, which increases BDNF, which activates TrkB and PI3K/Akt, which activates CREB, which further increases BDNF transcription.
This positive feedback mechanism may explain why P021's effects appear to be cumulative over time, with greater improvements in neurogenesis and cognition observed after longer treatment periods. It also raises interesting questions about whether the compound's benefits might persist after treatment cessation, as the elevated BDNF/CREB signaling could potentially sustain itself for some period. However, this hypothesis has not been formally tested, and the duration of any persistent effects remains unknown.
For researchers interested in how BDNF-related mechanisms compare across different nootropic peptides, our Peptide Hub provides detailed comparisons of neurotrophin-modulating compounds including Semax, which also upregulates BDNF but through a different upstream mechanism involving the melanocortin pathway.
Animal Cognitive Data
The cognitive effects of P021 have been evaluated across multiple animal models using a battery of well-validated behavioral tests. These studies collectively demonstrate that P021 can rescue cognitive deficits in models of Alzheimer's disease, Down syndrome, and normal aging, with effects that correlate with its neurogenic and neurotrophic molecular actions.
3xTg-AD Mouse Model: Alzheimer's Disease
The triple-transgenic Alzheimer's disease (3xTg-AD) mouse model harbors three human transgenes: presenilin 1 (PS1M146V), amyloid precursor protein (APPSwe), and tau (tauP301L). These mice develop both amyloid plaque and neurofibrillary tangle pathology in an age-dependent manner that roughly parallels the progression of human Alzheimer's disease, making them among the most widely used preclinical models for testing potential AD therapeutics.
Two major treatment paradigms have been tested with P021 in 3xTg-AD mice:
Early-Onset Treatment (3 Months of Age)
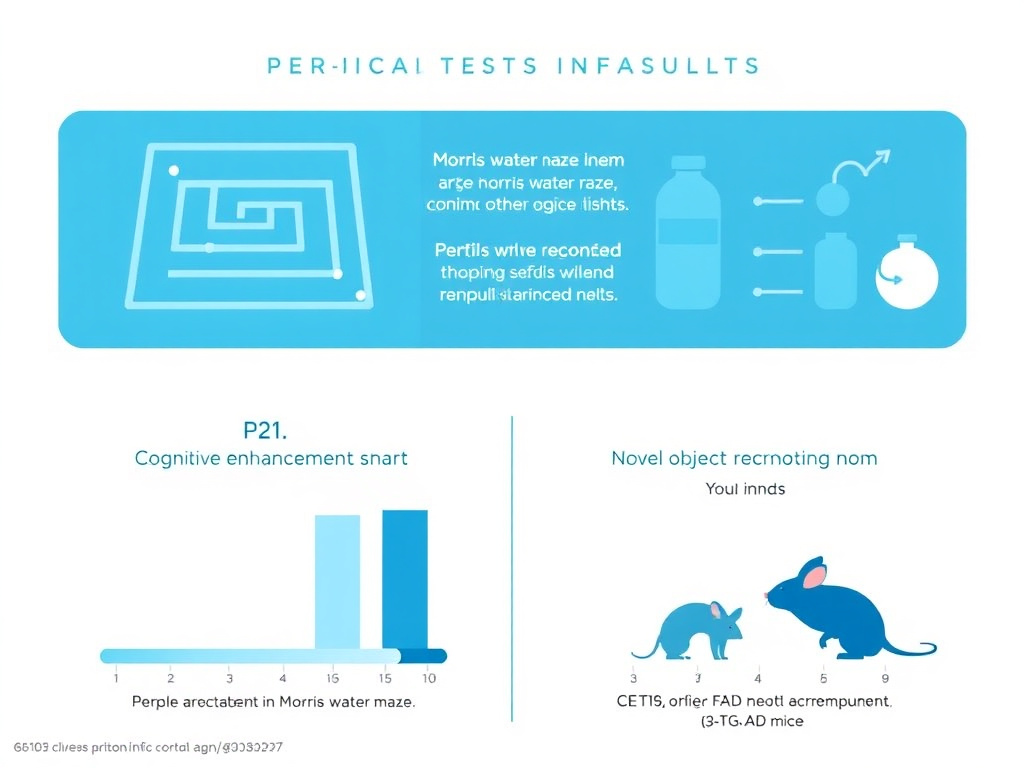
In studies conducted by Baazaoui and Iqbal (2017), P021 was administered orally at a dose of 60 nmol/g of feed beginning at 3 months of age, before the onset of any detectable pathology. Treatment continued for approximately 18 months. This prophylactic approach yielded the following results:
- Complete prevention of cognitive deficits as measured by Morris water maze (spatial learning and memory) and novel object recognition tests
- Significant reduction in amyloid-beta plaque burden and soluble amyloid-beta levels
- Marked reduction in hyperphosphorylated tau at multiple epitopes (AT8, PHF1, 12E8)
- Rescue of hippocampal neurogenesis to near wild-type levels
- Prevention of synaptic deficits (preserved synaptophysin and PSD-95 expression)
- No adverse effects on body weight, behavior, or general health over the entire 18-month treatment period
Late-Onset Treatment (9-10 Months of Age)
Perhaps more clinically relevant, P021 was also tested when treatment began at 9-10 months of age, a time point when amyloid pathology is already established in 3xTg-AD mice but tau pathology has not yet fully developed. This paradigm more closely approximates the clinical scenario in which treatment would begin after diagnosis, when brain pathology is already present. The results from studies by Kazim et al. (2014, 2017) showed:
- Rescue of spatial learning deficits in the Morris water maze at both 15-16 months and 21-22 months of age
- Restoration of hippocampal neurogenesis (increased Ki-67+ and DCX+ cells)
- Decreased levels of phosphorylated tau labeled with AT8, PHF1, and 12E8 antibodies
- Rescue of dendritic and synaptic deficits in CA1 hippocampal neurons
- Increased BDNF expression and enhanced CREB phosphorylation
- Inhibition of GSK-3 beta activity through increased Ser9 phosphorylation
The fact that P021 could reverse already-established cognitive deficits, not just prevent them, is a finding of considerable interest. It suggests that the compound's neurogenic and neurotrophic actions can compensate for existing neuronal damage and pathology, at least in this mouse model.
Hippocampal Neurogenesis Response to P21 Treatment
Data compiled from multiple preclinical studies. Values represent percentage of control group neurogenesis markers.
Ts65Dn Mouse Model: Down Syndrome
Down syndrome (trisomy 21) is associated with accelerated Alzheimer's-like pathology and cognitive impairment, making it a relevant model for testing neurotrophic interventions. The Ts65Dn mouse carries a partial trisomy of chromosome 16, which includes orthologues of many genes on human chromosome 21, and displays cognitive deficits, reduced neurogenesis, and age-dependent Alzheimer-like changes.
Bhatt et al. studied the effects of P021 treatment during prenatal to early postnatal development in Ts65Dn mice. Pregnant dams received P021 in their feed (60 nmol/g), and treatment continued through weaning. The study found that this early intervention:
- Rescued developmental delay in Ts65Dn pups
- Prevented Alzheimer's-like hippocampus-dependent memory impairments that typically emerge in adult Ts65Dn mice
- Did not induce changes in body weight, anxiety-like behavior, or general activity levels
- Enhanced BDNF signaling in the hippocampus
These results in a developmental disorder model broaden the potential therapeutic applications of P021 beyond late-onset neurodegenerative disease.
Aged Fischer Rats: Cognitive Aging
Normal cognitive aging, independent of any disease process, is associated with declining neurogenesis, reduced BDNF levels, and progressive memory impairment. To determine whether P021 could address age-related cognitive decline, Bolognin et al. (2014) tested the compound in 22- to 24-month-old Fischer 344 rats, the equivalent of approximately 65-70 years in human terms.
Chronic oral administration of P021 for several months significantly reduced the age-dependent decline in learning and memory performance. Treated aged rats showed:
- Improved performance on spatial learning tasks compared to age-matched controls
- Increased hippocampal neurogenesis (more BrdU/NeuN double-labeled cells)
- Elevated BDNF protein levels in the hippocampus
- Reduced tau hyperphosphorylation at disease-relevant epitopes
These findings in a normal aging model, rather than a genetic disease model, suggest that P021's cognitive benefits extend to the general population of aging individuals, not just those with specific pathological conditions. The implications for human cognitive aging are intriguing, though the usual caveats about translating rodent findings to humans apply with full force.
CDKL5 Deficiency Disorder
A 2024 study by Galvani et al. expanded the investigation of P021 into CDKL5 deficiency disorder (CDD), a rare neurodevelopmental condition caused by mutations in the CDKL5 gene. Using both in vitro (human CDKL5-mutant neurons derived from induced pluripotent stem cells) and in vivo (Cdkl5 knockout mice) models, the researchers found that P021 increased BDNF expression and enhanced neurogenesis. However, the therapeutic effects were partial, with some endpoints showing improvement while others did not reach significance. This study represents the most recent expansion of P021 research into new disease models and suggests that while the compound's BDNF-enhancing mechanism has broad applicability, its efficacy may vary by condition.
Summary of Behavioral Findings Across Models
| Model | Age at Treatment Start | Duration | Cognitive Outcome | Neurogenesis Effect |
|---|---|---|---|---|
| 3xTg-AD (early) | 3 months | 18 months | Prevention of deficits | Maintained at WT levels |
| 3xTg-AD (late) | 9-10 months | 6-12 months | Rescue of deficits | Restored to near WT levels |
| Ts65Dn (Down syndrome) | Prenatal | Through weaning | Prevention of impairments | Enhanced |
| Aged Fischer rats | 22-24 months | Several months | Reduced age-related decline | Increased BrdU+/NeuN+ cells |
| Cdkl5 KO mice (CDD) | Various | Various | Partial improvement | Increased (partial) |
For those interested in exploring other peptides with documented cognitive effects in animal models, our research library includes detailed reports on Semax and Dihexa, both of which enhance cognition through complementary but distinct mechanisms.
Alzheimer's Disease Model Research
Alzheimer's disease (AD) represents the primary therapeutic target for P021 development, and the compound has been more extensively studied in AD models than in any other disease context. The research spans over a decade and includes multiple studies using the 3xTg-AD mouse model, with treatment paradigms ranging from early prevention to late-stage intervention.
Understanding P021's Multi-Target Approach to AD Pathology
Most failed Alzheimer's drug candidates have targeted a single pathological mechanism, typically amyloid-beta production or aggregation. The "amyloid hypothesis" has dominated AD drug development for decades, yet dozens of anti-amyloid therapies have failed in clinical trials. This has led to growing recognition that effective AD treatment will likely require addressing multiple pathological mechanisms simultaneously.
P021 is noteworthy because it targets multiple aspects of AD pathology through a single upstream mechanism (LIF inhibition and BDNF upregulation):
- Tau hyperphosphorylation: Reduced through GSK-3 beta inhibition via the BDNF/TrkB/PI3K/Akt pathway
- Amyloid-beta pathology: Reduced plaque burden and soluble A-beta levels, likely through enhanced clearance mechanisms and reduced production
- Neurogenesis impairment: Restored hippocampal neurogenesis through direct neurogenic stimulation
- Synaptic loss: Rescued synaptic protein expression and dendritic morphology through BDNF-mediated trophic support
- Cognitive decline: Reversed learning and memory deficits through combined neurogenic and neuroprotective actions
Tau Pathology Reduction
The reduction of tau hyperphosphorylation by P021 has been documented at multiple phosphorylation sites using well-characterized antibodies. In 3xTg-AD mice treated with P021 starting at 9-10 months of age, significant reductions were observed after 6 months of treatment in the following tau epitopes:
- AT8 (Ser202/Thr205): This epitope is an early marker of tau pathology and is routinely used for Braak staging in human AD brains. P021 significantly reduced AT8-positive tau in the hippocampus and cortex.
- PHF1 (Ser396/Ser404): These phosphorylation sites are associated with paired helical filament formation and are considered markers of more advanced tau pathology. P021 reduced PHF1 immunoreactivity.
- 12E8 (Ser262/Ser356): Phosphorylation at these sites within the microtubule-binding repeats of tau directly impairs tau's ability to bind and stabilize microtubules. P021 reduced 12E8-positive tau.
The reduction in tau hyperphosphorylation was mechanistically linked to increased inhibitory phosphorylation of GSK-3 beta at Ser9, confirming the proposed BDNF/TrkB/PI3K/Akt/GSK-3 beta pathway. Total tau levels were not significantly changed by P021 treatment, indicating that the compound specifically reduces pathological phosphorylation rather than altering tau expression.
Amyloid-Beta Reduction
While P021 was not designed as an anti-amyloid agent, its chronic administration in 3xTg-AD mice produced notable reductions in amyloid-beta pathology. Both extracellular amyloid plaque burden and intracellular soluble amyloid-beta levels were reduced in treated animals compared to vehicle controls. The mechanism behind this anti-amyloid effect is not entirely clear but may involve:
- Enhanced amyloid-beta clearance through BDNF-mediated activation of proteolytic enzymes (neprilysin, insulin-degrading enzyme)
- Reduced amyloid precursor protein (APP) processing through the amyloidogenic pathway
- Improved neuronal health and proteostasis, reducing the stress-related production of amyloid-beta
The dual reduction of both tau and amyloid pathology by a single compound is relatively unusual and may reflect the interconnected nature of these pathological cascades. GSK-3 beta, which is inhibited by P021 through the BDNF pathway, has been shown to regulate both tau phosphorylation and amyloid-beta production, providing a mechanistic basis for the compound's effects on both pathologies.
Retinal Neurodegeneration (AMD-like Pathology)
An interesting extension of P021 research into Alzheimer's-related pathology was conducted by Kazim et al. (2019), who examined whether P021 could protect against age-related macular degeneration (AMD)-like pathology. Both aging and Alzheimer's disease are risk factors for AMD, and the retina shares developmental origins and molecular machinery with the brain.
In aged rats and 3xTg-AD mice, P021 treatment inhibited AMD-like pathological features in the retina, including reduced drusen-like deposits, preserved retinal layer thickness, and maintained visual function. This finding suggests that P021's neurotrophic effects extend beyond the brain to other neural tissues, expanding its potential therapeutic applications.
The Disease Modification Question
A key question for any Alzheimer's therapeutic candidate is whether it provides disease modification, meaning it slows or halts the underlying disease process, as opposed to symptomatic relief. The evidence from P021 preclinical studies supports a disease-modifying mechanism:
- P021 reduces pathological hallmarks of AD (tau tangles, amyloid plaques), not just symptoms
- The compound promotes structural repair (neurogenesis, synaptogenesis, dendritic remodeling)
- Benefits accumulate and persist over long treatment periods rather than diminishing (tolerance development)
- Treatment started after pathology onset still produces significant improvements
Important Limitations
All Alzheimer's disease data for P021 comes from animal models. The 3xTg-AD mouse, while widely used, does not fully recapitulate human AD. Mice do not develop the same pattern of neuronal loss, their tau pathology is driven by a mutant transgene rather than wild-type tau, and the time course of disease progression differs substantially from the human condition. Many compounds that show efficacy in AD mouse models have failed in human clinical trials. P021's effects in humans remain entirely unknown, and no clinical trials have been initiated as of 2026.
For a broader perspective on peptide-based approaches to neurodegenerative disease, see our comprehensive Biohacking Hub, which covers neuroprotective strategies across multiple compound classes.
Comparison to Cerebrolysin, Semax, and Dihexa
P21 occupies a unique position among nootropic peptides. To understand its distinctive features and limitations, it helps to compare it directly with three closely related compounds: Cerebrolysin (from which P21's design was partly inspired), Semax (another neurotrophin-modulating peptide), and Dihexa (a potent neurogenic peptide targeting a different growth factor pathway).
P21 vs. Cerebrolysin
Cerebrolysin is a porcine brain-derived peptide preparation that has been used clinically in parts of Europe and Asia for decades, primarily for stroke recovery and traumatic brain injury. It contains a complex mixture of low-molecular-weight neuropeptides and free amino acids that includes fragments of CNTF, BDNF, NGF, GDNF, and other neurotrophic factors.
| Feature | P21 (P021) | Cerebrolysin |
|---|---|---|
| Composition | Single synthetic peptide (Ac-DGGLAG-NH2) | Complex mixture of 100s of peptides |
| Molecular Weight | 578.3 Da | Variable (< 10 kDa mixture) |
| Primary Mechanism | LIF inhibition, BDNF upregulation | Multiple neurotrophic pathways |
| Oral Bioavailability | Yes (confirmed in animals) | No (requires IV/IM injection) |
| BBB Penetration | Yes (adamantane modification) | Yes (small peptides in mixture) |
| Reproducibility | Fully synthetic, batch-to-batch consistent | Biological extract, variable |
| Human Clinical Data | None | Multiple clinical trials (stroke, TBI, AD) |
| Regulatory Status | Research compound only | Approved in some countries (not US/UK) |
| Administration Route | Oral, intranasal, subcutaneous | Intravenous or intramuscular |
| Cost | Research peptide pricing | Medical-grade pricing |
P21's advantages over Cerebrolysin include its defined molecular identity (enabling precise dosing, quality control, and mechanistic study), oral bioavailability (eliminating the need for injections), and synthetic production (ensuring batch-to-batch consistency). However, Cerebrolysin has a significant advantage in clinical evidence: it has been tested in human clinical trials for multiple indications and is approved for clinical use in several countries. P21 has no human data whatsoever.
It's also worth noting that Cerebrolysin's therapeutic activity may derive from its combination of multiple neurotrophic peptide fragments acting on different pathways simultaneously. P21's focused mechanism (primarily BDNF upregulation through LIF inhibition) may not capture all the therapeutic effects of the complex mixture. On the other hand, P21's selectivity avoids the "dirty drug" problem, where complex mixtures can produce unpredictable interactions and variable effects.
P21 vs. Semax
Semax is a synthetic heptapeptide analog of adrenocorticotropic hormone (ACTH) fragments 4-7 (Met-Glu-His-Phe) with a Pro-Gly-Pro tripeptide extension that enhances metabolic stability. Developed in Russia, Semax has been approved for clinical use in Russia and several CIS countries for stroke, cognitive disorders, and optic nerve disease.
| Feature | P21 (P021) | Semax |
|---|---|---|
| Origin | CNTF epitope (residues 148-151) | ACTH(4-7) analog |
| Primary Target | LIF/BDNF axis | Melanocortin system |
| BDNF Effect | Upregulation (via LIF inhibition) | Upregulation (via melanocortin pathway) |
| Neurogenesis | Strong (dentate gyrus) | Moderate |
| Onset of Action | Weeks to months (cumulative) | Days to weeks (faster onset) |
| Primary Indication | Neurodegeneration, cognitive aging | Stroke, cognitive disorders |
| Human Data | None | Limited clinical data (Russia) |
| Route | Oral, intranasal, subcutaneous | Intranasal (primary) |
| Anti-tau Effect | Yes (GSK-3 beta inhibition) | Not demonstrated |
| Anti-amyloid Effect | Yes (indirect) | Neuroprotective but different mechanism |
Semax and P21 both upregulate BDNF, but through completely different upstream mechanisms. Semax acts through the melanocortin receptor system and influences dopamine and serotonin neurotransmitter levels, providing more rapid cognitive effects. P21's effects are more targeted toward structural neuroplasticity and disease modification. In theory, the two compounds could be complementary because they converge on BDNF enhancement through independent pathways.
P21 vs. Dihexa
Dihexa (N-hexanoyl-Tyr-Ile-(6)-aminohexanoic amide) is a synthetic hexapeptide analog of angiotensin IV that was developed at Washington State University. It acts by potentiating hepatocyte growth factor (HGF) and its receptor c-Met, a pathway involved in neurogenesis, synaptogenesis, and neuronal survival.
| Feature | P21 (P021) | Dihexa |
|---|---|---|
| Primary Target | LIF/BDNF/TrkB pathway | HGF/c-Met pathway |
| Neurogenesis | Strong (dentate gyrus) | Strong (multiple regions) |
| Synaptogenesis | Yes (BDNF-mediated) | Yes (HGF/c-Met-mediated) |
| Relative Potency | Active at nmol/g doses orally | Active at picomolar concentrations |
| Oral Bioavailability | Yes | Yes |
| Anti-tau Effect | Yes | Not established |
| AD-Specific Research | Extensive (3xTg-AD model) | Limited |
| Human Data | None | None |
| Safety Concern | Minimal in animal studies | Potential oncogenicity (HGF/c-Met) |
Dihexa is sometimes described as the most potent nootropic peptide ever discovered, based on its activity at picomolar concentrations. However, potency alone does not determine clinical utility. P21 has a more extensive safety record in long-term animal studies and a better-characterized disease-modification profile in AD models. Dihexa's activation of the HGF/c-Met pathway raises theoretical concerns about oncogenic potential, since this pathway is frequently dysregulated in cancers, though no tumors have been reported in animal studies.
The two compounds target different growth factor pathways (BDNF/TrkB vs. HGF/c-Met), which means they could potentially be used together to activate complementary neuroplasticity mechanisms. This stacking approach is discussed further in the Stacking section below.
Choosing Between Nootropic Peptides
Each of these compounds has distinct strengths. P21 offers the most targeted Alzheimer's disease research and the cleanest long-term safety data. Semax provides the fastest onset of cognitive effects and has some human clinical data. Dihexa offers the highest potency and strongest synaptogenic activity. Cerebrolysin has the most extensive human clinical trial data. None of these compounds has been approved by the FDA, and all represent experimental research tools. For personalized guidance, consider using our Free Assessment tool.
Dosing Protocols and Administration
P21 dosing information comes from two sources: the controlled doses used in published animal research and the anecdotal protocols reported within the biohacker and nootropic communities. It is essential to distinguish between these two very different evidence bases, as only the animal data carries scientific weight.
Research Compound Disclaimer
P21 has not been tested in human clinical trials. No human dosing data exists from controlled studies. All dosing information presented here is derived from animal research or anecdotal community reports. P21 is sold as a research compound, not for human consumption. Any use in humans is entirely self-directed and carries unknown risks. Consult a qualified healthcare provider before considering any experimental compound.
Animal Research Doses
In the published preclinical literature, P021 has been administered primarily through two routes:
Oral Administration (Feed-Based)
The most common dosing method in published research has been incorporation of P021 into animal feed at a concentration of 60 nmol per gram of feed. This provides chronic, continuous exposure through normal eating behavior and has been used in the long-term studies in 3xTg-AD mice (up to 18 months), aged Fischer rats, and Ts65Dn mice. The exact daily intake varies based on food consumption but provides a steady-state concentration sufficient to achieve CNS penetration and biological effects.
Based on typical mouse food intake (approximately 3-5 grams per day for a 25-30 gram mouse), this translates to roughly 180-300 nmol per day, or approximately 104-173 micrograms per day per mouse. Allometric scaling from mouse to human doses is complex and imprecise, but rough estimates using body surface area conversion suggest a human-equivalent dose in the range of 0.5-2 mg per day. These are rough calculations and should be treated with extreme caution.
Subcutaneous Injection
Some studies have used subcutaneous injection for more precise dose control. This route provides consistent systemic delivery and avoids first-pass metabolism. The doses used have varied by study and model but have generally been in the range of 30-100 nmol/mouse/day.
Community-Reported Protocols
Within the nootropic community, P21 is commonly used via two administration routes. These protocols are based entirely on anecdotal reports and have not been validated in controlled studies.
Intranasal Administration
The most popular route among biohackers, intranasal administration bypasses the blood-brain barrier by allowing direct transport along the olfactory nerve pathway to the brain. Community-reported doses typically range from 500 mcg to 1 mg daily as a starting dose, with some users increasing to 2-4 mg for stronger acute effects.
Reported intranasal protocol details:
- Reconstitute lyophilized P21 with bacteriostatic water or sterile saline
- Administer to clean nasal passages
- Tilt head slightly back during administration
- Avoid blowing the nose for 10-15 minutes after dosing
- Alternate nostrils between doses
- Take at the same time each day for consistent neurotrophic stimulation
Subcutaneous Injection
Some users prefer subcutaneous injection for more precise dosing and consistent absorption. Community-reported doses range from 100-500 mcg daily, typically administered in cycles of 4-6 weeks followed by a break period.
Reported subcutaneous protocol details:
- Reconstitute with bacteriostatic water
- Common injection sites: lower abdomen, front of thighs, back of upper arm
- Use insulin syringes for precise measurement
- Rotate injection sites to prevent local tissue irritation
- Typical cycle: 4-6 weeks on, 2-4 weeks off
For help calculating precise volumes and concentrations from reconstituted peptides, our Dosing Calculator can simplify the math.
Onset and Duration of Effects
Based on the animal research, P21's neurogenic effects are cumulative and build over weeks to months. Users should not expect immediate cognitive changes. The published studies showed cognitive improvements emerging after weeks of treatment, with continued improvement over months. This is consistent with the compound's mechanism: neurogenesis is a slow process that requires weeks for new neurons to mature and integrate into functional circuits.
Some community reports describe subtle improvements in mood, focus, and mental clarity beginning within the first one to two weeks, potentially attributable to the more rapid BDNF-mediated effects on synaptic plasticity before the neurogenic effects become apparent. These reports are subjective and uncontrolled, so they should be interpreted cautiously.
Cycling Considerations
The animal research employed continuous daily treatment for extended periods (months to over a year) without evidence of tolerance development or diminishing returns. In fact, the data suggest that longer treatment periods produce greater benefits. This contrasts with the cycling approach (on-off periods) commonly recommended in the nootropic community.
The rationale for cycling in community protocols is based on the general principle that receptor systems can desensitize with chronic stimulation. However, P021's mechanism (competitive inhibition of LIF signaling) may not be susceptible to traditional receptor desensitization in the same way that direct receptor agonists might be. The absence of tolerance in 18-month animal studies supports this interpretation, though the question has not been formally studied.
| Parameter | Intranasal (Community) | Subcutaneous (Community) | Oral (Animal Research) |
|---|---|---|---|
| Starting Dose | 500 mcg - 1 mg/day | 100-500 mcg/day | 60 nmol/g feed (~100-175 mcg/day mouse) |
| Higher Dose | 2-4 mg/day | 500 mcg/day | Same dose throughout studies |
| Cycle Length | 4-8 weeks | 4-6 weeks | Continuous (up to 18 months) |
| Onset | 1-2 weeks (subjective) | 1-2 weeks (subjective) | Weeks to months (objective measures) |
| Evidence Level | Anecdotal only | Anecdotal only | Peer-reviewed animal data |
Stacking P21 with Other Nootropic Compounds
The concept of "stacking," combining multiple compounds to achieve enhanced effects through complementary mechanisms, is popular in the nootropic community. P21's well-defined mechanism of action (LIF inhibition leading to BDNF upregulation) makes it theoretically compatible with compounds that enhance cognition through different pathways. However, it's critical to acknowledge that no stacking combination involving P21 has been tested in any published study. All stacking suggestions are based on mechanistic reasoning and anecdotal reports, not empirical evidence.
Theoretical Stacking Rationale
P21 + Semax
Both Semax and P21 upregulate BDNF, but through entirely different upstream mechanisms. Semax acts via the melanocortin receptor system and modulates dopamine and serotonin signaling, while P21 acts through LIF inhibition. In theory, combining these two pathways could produce additive or amplified BDNF elevation. Semax also provides more rapid-onset cognitive effects (improved focus, processing speed), which could complement P21's slower-building neurogenic benefits.
P21 + Dihexa
Dihexa activates the HGF/c-Met pathway, which is entirely separate from P21's BDNF/TrkB pathway. Together, these compounds would theoretically activate two major neuroplasticity signaling cascades simultaneously. P21 promotes new neuron birth, while Dihexa strengthens synaptic connections between existing neurons, potentially creating a complementary "build and connect" strategy.
P21 + Selank
Selank is an anxiolytic peptide that modulates GABA-ergic transmission and has immunomodulatory properties. Its anxiolytic effects could complement P21's neurogenic actions by reducing stress-related suppression of neurogenesis. Chronic stress is one of the most potent inhibitors of hippocampal neurogenesis, so reducing anxiety and stress signaling could theoretically amplify P21's neurogenic effects.
P21 + NAD+ Precursors
NAD+ and its precursors (NMN, NR) support cellular energy metabolism and sirtuin activation. Since neurogenesis is an energy-intensive process, adequate NAD+ levels could support the metabolic demands of new neuron production stimulated by P21. Additionally, NAD+-dependent sirtuins (particularly SIRT1) have been shown to regulate BDNF expression and hippocampal plasticity.
P21 + GHK-Cu
GHK-Cu (copper peptide) has been shown to modulate gene expression of multiple growth factors and has anti-inflammatory properties. Its ability to reduce chronic inflammation could theoretically support P21's neurogenic actions by creating a less hostile microenvironment for newly born neurons.
P21 + Pinealon
Pinealon is a tripeptide (Glu-Asp-Arg) that has been studied for its neuroprotective properties, particularly in the context of oxidative stress and excitotoxicity. Combining Pinealon's neuroprotective effects with P21's neurogenic actions could theoretically provide both protection of existing neurons and generation of new ones.
P21 + Epithalon
Epithalon (Epitalon) is a tetrapeptide that stimulates telomerase activity and has been associated with anti-aging effects. Since neurogenesis declines with age partly due to neural stem cell senescence, Epithalon's telomere-protective effects could theoretically support the long-term viability of the neural stem cell population that P21 stimulates.
Stacking Safety Considerations
No published research has tested any P21 stacking combination in animals or humans. Combining experimental peptides multiplies the unknowns regarding safety, drug interactions, and unpredictable effects. The theoretical rationale for stacking, while mechanistically logical, does not constitute evidence of safety or efficacy. Users who choose to combine peptides do so at their own risk and should proceed with extreme caution, ideally under medical supervision.
Safety Profile
P21's safety profile is one of its more encouraging characteristics, at least within the context of preclinical animal studies. The compound was specifically designed to avoid the adverse effects of its parent molecule CNTF, and the available data suggests this design goal was achieved. However, the absence of any human safety data represents a significant gap in the evidence base.
Animal Safety Data
The most comprehensive safety assessment of P021 comes from the long-term studies in 3xTg-AD mice, where the compound was administered orally at 60 nmol/g of feed for up to 18 months continuously. This represents the equivalent of roughly 40-50 years of human treatment, adjusting for the compressed lifespan of mice. Key safety observations from these studies include:
- Body weight: No significant changes in body weight were observed throughout the treatment period. This is a direct contrast to full-length CNTF, which causes severe anorexia and weight loss in both animals and humans. The absence of weight loss confirms that P021 does not activate the IL-6R alpha/LIFR beta/gp130 receptor complex responsible for CNTF's catabolic effects.
- Tumor formation: No tumors or neoplastic changes were observed in any treated animal over the 18-month period. This is relevant because neurotrophic factors and their mimetics have theoretical oncogenic potential due to their pro-growth signaling effects.
- Pain and distress: No signs of pain, hyperalgesia, or discomfort were observed in treated animals. Again, this contrasts with full-length CNTF, which causes severe muscle cramps and pain.
- Behavior: P021 treatment did not alter anxiety-like behavior (elevated plus maze), general locomotor activity, or exploratory behavior. This indicates that the compound does not produce sedation, hyperactivity, or anxiogenic effects.
- Organ histopathology: No histopathological abnormalities were reported in major organs of treated animals.
In the Ts65Dn mouse model of Down syndrome, P021 administered during the prenatal-to-early-postnatal period did not affect maternal health, litter size, pup birth weight, or developmental milestones (other than the intended rescue of developmental delay).
Why P21 Avoids CNTF's Side Effects
The selectivity of P021's mechanism explains its favorable safety profile. Full-length CNTF's adverse effects, particularly anorexia and weight loss, are mediated through its interaction with the IL-6R alpha/LIFR beta/gp130 receptor complex, which activates inflammatory signaling pathways in peripheral tissues. The CNTF side effects are thought to result from activation of this alternative receptor complex rather than the primary CNTF receptor alpha.
P021, being a small peptide derived from only a portion of the CNTF molecule, does not interact with the full receptor complex in the same way. Its action is more selective, primarily affecting the LIF signaling pathway in the brain to modulate BDNF expression. This selectivity eliminates the peripheral inflammatory and catabolic effects while preserving the central neurotrophic actions.
Theoretical Safety Concerns
Despite the encouraging animal data, several theoretical safety concerns warrant discussion:
- Long-term neurogenesis stimulation: While increased neurogenesis is generally considered beneficial, there are theoretical concerns about chronic stimulation of neural stem cell proliferation. Could this eventually deplete the stem cell pool? Could aberrant neurogenesis contribute to epileptogenesis? These questions have not been addressed in the P021 literature.
- BDNF overexpression: While BDNF deficiency contributes to neurodegeneration, excessive BDNF levels have been associated with epileptiform activity and pain hypersensitivity in some experimental contexts. Whether P021 could produce supra-physiological BDNF levels with chronic use is unknown.
- GSK-3 beta inhibition: GSK-3 beta regulates numerous cellular processes beyond tau phosphorylation, including glycogen metabolism, cell cycle progression, and Wnt signaling. Chronic inhibition could theoretically have metabolic or developmental consequences, though no such effects were observed in the animal studies.
- Species differences: The favorable safety profile in mice may not translate directly to humans. Human neurobiology differs from rodent neurobiology in numerous ways that could affect both efficacy and safety.

Known Interactions and Contraindications
No formal drug interaction studies have been conducted with P021. Based on its mechanism of action, the following theoretical interactions should be considered:
- Lithium: Lithium is a GSK-3 beta inhibitor. Combining it with P021 (which also reduces GSK-3 beta activity indirectly) could produce excessive GSK-3 beta inhibition.
- Antiepileptic drugs: Given the theoretical concern about BDNF-related seizure threshold changes, users of antiepileptic medications should exercise particular caution.
- Other neurotrophic compounds: Combining P021 with other BDNF-enhancing agents could produce additive increases in BDNF levels, with unknown consequences.
- Immunosuppressants: P021's effects on LIF and JAK-STAT signaling could theoretically interact with immunosuppressive drugs that target these pathways.
Pregnant and breastfeeding individuals should avoid P021 entirely. While the Down syndrome prevention study used P021 during pregnancy in mice with positive results, the effects of the compound on human fetal development are completely unknown.
Emerging Research Applications and Future Directions
Traumatic Brain Injury
P021's neurogenic and neuroprotective properties make it a logical candidate for traumatic brain injury (TBI) research. TBI is characterized by acute neuronal death followed by secondary cascading injury processes, including inflammation, excitotoxicity, and progressive neurodegeneration. BDNF levels drop significantly after TBI, and restoring BDNF signaling has been associated with improved outcomes in animal models of brain trauma.
While no published studies have specifically tested P021 in TBI models, the compound's ability to upregulate BDNF, promote neurogenesis, and protect against neurodegeneration in AD models provides a strong mechanistic rationale for such studies. Given the significant unmet need in TBI treatment (currently, there are no approved pharmacological treatments that improve long-term outcomes after TBI), this represents a promising research avenue.
Age-Related Macular Degeneration
The study by Kazim et al. (2019) showing that P021 inhibited AMD-like pathology in aged rats and 3xTg-AD mice opens up the possibility of ophthalmic applications. The retina is a neural tissue that shares many molecular pathways with the brain, and BDNF signaling is important for retinal ganglion cell survival and photoreceptor maintenance. If P021's neuroprotective effects extend to retinal neurons, it could have applications in AMD, glaucoma, and other neurodegenerative eye diseases.
Depression and Mood Disorders
The "neurotrophic hypothesis of depression" posits that reduced BDNF levels contribute to the pathophysiology of major depressive disorder. Antidepressant medications, including SSRIs and SNRIs, increase BDNF expression in the hippocampus with chronic treatment, and this BDNF increase is believed to mediate their therapeutic effects at least in part. Similarly, antidepressant effects have been linked to hippocampal neurogenesis.
P021's ability to strongly increase both BDNF levels and hippocampal neurogenesis makes it a theoretical candidate for antidepressant research. No studies have specifically tested P021 in animal models of depression, but the mechanistic rationale is compelling.
Phanes Biotech and Clinical Translation
Phanes Biotech, the company co-founded by Dr. Khalid Iqbal (who serves as Chief Scientific Officer), is working to advance P021 toward human clinical trials. The company's focus is on developing P021 as a disease-modifying therapy for Alzheimer's disease and related neurodegenerative conditions. Key factors that support clinical translatability include:
- Well-defined molecular target and mechanism of action
- Oral bioavailability (rare for peptide therapeutics)
- Excellent preclinical safety record (18 months in mice)
- Disease modification rather than symptomatic treatment
- Multi-target effects through a single upstream mechanism
- Low molecular weight and synthetic producibility
However, the path from promising preclinical compound to approved drug is long and fraught with challenges. The vast majority of compounds that show efficacy in AD mouse models fail in human clinical trials. Regulatory requirements for CNS-active compounds are particularly stringent, and the clinical trial pathway for disease-modifying AD therapies typically requires large, long-duration studies with hundreds or thousands of patients.
Pharmacokinetics and Bioavailability
P021's pharmacokinetic profile is one of its most distinguishing features among peptide therapeutics. Most peptides are rapidly degraded in the gastrointestinal tract and have poor oral bioavailability, necessitating injection. P021's adamantane modification was specifically designed to overcome these limitations, and the available data suggests it succeeded.
Gastrointestinal Stability
In vitro stability studies demonstrated that P021 retains over 90% of its structural integrity after 30 minutes in artificial gastric juice at 37 degrees Celsius. In artificial intestinal fluid under the same conditions, stability exceeded 97% after 2 hours. These values are exceptionally high for a peptide compound and are attributed to the steric protection provided by the C-terminal adamantylated glycine and the N-terminal acetylation, which together shield the peptide bonds from attack by both endopeptidases and exopeptidases.
Blood-Brain Barrier Penetration
The lipophilic adamantane moiety dramatically increases the compound's partition coefficient, enhancing its ability to cross cell membranes, including the blood-brain barrier. BBB penetration has been confirmed by detection of intact P021 in brain tissue following oral and peripheral administration in mice. The efficiency of BBB crossing has not been precisely quantified (e.g., as a percentage of administered dose reaching the CNS), but the biological activity of oral P021 on brain endpoints (neurogenesis, BDNF levels, cognitive performance) confirms functionally relevant CNS exposure.
Plasma Half-Life and Distribution
P021 has a plasma half-life exceeding 3 hours in mice, which is unusually long for a peptide of this size. For comparison, most unmodified tetrapeptides have plasma half-lives measured in minutes due to rapid enzymatic degradation. The extended half-life supports once-daily dosing in the animal studies and is consistent with the observed chronic efficacy of oral administration.
Tissue distribution studies suggest that P021 reaches therapeutic concentrations in the hippocampus and cortex following oral administration, with sufficient exposure to activate the LIF/BDNF signaling cascade. Detailed pharmacokinetic parameters such as volume of distribution (Vd), area under the curve (AUC), and clearance (CL) have not been published for the compound.
Intranasal Pharmacokinetics
While intranasal administration is the most popular route among users in the nootropic community, it has not been the primary route studied in published research. Intranasal delivery theoretically bypasses the blood-brain barrier entirely by allowing direct transport along the olfactory nerve and trigeminal nerve pathways to the brain. This could provide higher CNS concentrations relative to peripheral exposure compared to oral or subcutaneous routes. However, formal pharmacokinetic studies comparing intranasal versus oral P021 bioavailability have not been published.
| PK Parameter | Value | Method/Species |
|---|---|---|
| Gastric Stability (30 min) | > 90% | In vitro, 37C |
| Intestinal Stability (2 hr) | > 97% | In vitro, 37C |
| Plasma Half-life | > 3 hours | In vivo, mouse |
| Oral Bioavailability | Confirmed (not quantified %) | In vivo, mouse (functional endpoint) |
| BBB Penetration | Confirmed | In vivo, mouse (tissue detection) |
| Molecular Weight | 578.3 Da | Calculated |
Molecular Biology: CNTF Receptor Signaling and P21's Selectivity
To fully appreciate P21's mechanism, it's useful to understand the signaling biology of CNTF and the IL-6 cytokine family in greater detail.
CNTF Receptor Complexes
CNTF signals through a complex receptor system that can form multiple configurations. The canonical CNTF signaling complex consists of CNTF receptor alpha (CNTFR alpha), leukemia inhibitory factor receptor beta (LIFR beta), and glycoprotein 130 (gp130). CNTF first binds to CNTFR alpha, and this binary complex then recruits LIFR beta and gp130, forming a hexameric signaling complex. The intracellular domains of LIFR beta and gp130 activate Janus kinases (JAKs), which phosphorylate signal transducer and activator of transcription 3 (STAT3), triggering its dimerization, nuclear translocation, and activation of target gene transcription.
CNTF can also signal through an alternative receptor configuration involving IL-6R alpha, LIFR beta, and gp130. This alternative signaling is responsible for many of CNTF's peripheral adverse effects, including activation of the acute phase response, fever, cachexia (weight loss and muscle wasting), and pain. The differential tissue distribution of CNTFR alpha (primarily neural) versus IL-6R alpha (widely expressed in peripheral tissues) explains why CNTF has both beneficial neural effects and harmful systemic effects.
P21's Selective Mechanism
P021 was designed to capture the neurotrophic activity of CNTF while avoiding the adverse effects mediated through the alternative receptor complex. The peptide competitively inhibits LIF signaling through the JAK-STAT3 pathway, but does so with selectivity that spares the alternative inflammatory pathway.
The key molecular insight is that P021 does not function as a traditional CNTF receptor agonist. Instead, it acts by modulating the balance of signaling through the CNTF/LIF receptor system. By competitively inhibiting LIF's tonic suppressive effect on neurogenesis and BDNF expression, P021 effectively "releases the brake" on neurotrophic signaling without directly activating the receptor complexes responsible for adverse effects. This indirect mechanism of action is fundamentally different from administering recombinant CNTF, which indiscriminately activates all CNTF receptor configurations.
STAT3 and Neural Stem Cell Biology
The JAK-STAT3 pathway, which P021 modulates through LIF inhibition, plays a complex role in neural stem cell biology. In the adult hippocampal neurogenic niche, STAT3 signaling can either promote or inhibit neurogenesis depending on the context, the specific upstream activator, and the developmental stage of the neural progenitor cell.
LIF-mediated STAT3 activation tends to maintain neural stem cells in a quiescent, undifferentiated state, which limits the production of new neurons. By inhibiting this LIF-driven STAT3 activation, P021 allows neural stem cells to exit quiescence and enter the proliferative phase, where they can give rise to new neuroblasts that will eventually mature into functional neurons. Simultaneously, the increase in BDNF expression provides trophic support for the survival and maturation of these newly born cells.
P21 in the Broader Nootropics Landscape
The nootropic peptide field has expanded considerably in recent years, with compounds targeting a variety of neurobiological mechanisms. Understanding where P21 fits within this broader landscape helps contextualize its unique value proposition and limitations.
Neurotrophin-Modulating Peptides
P21 belongs to a class of compounds that modulate neurotrophin signaling, with BDNF as the primary downstream effector. Other peptides in this class include:
- 7,8-Dihydroxyflavone (7,8-DHF): A small molecule TrkB agonist that directly activates the BDNF receptor without increasing BDNF expression itself. Unlike P21, which increases endogenous BDNF, 7,8-DHF bypasses BDNF entirely and directly stimulates the receptor. This distinction matters because endogenous BDNF has regulatory features (activity-dependent release, spatial and temporal specificity) that direct receptor agonism lacks.
- NSI-189: A neurogenic compound developed by Neuralstem that stimulates hippocampal neurogenesis through a mechanism that may involve BDNF modulation. Unlike P21, NSI-189 has been tested in Phase II clinical trials for major depressive disorder, providing human safety data.
- Cerebrolysin: As discussed, the complex peptide mixture from which P21's design was partly inspired. Cerebrolysin modulates multiple neurotrophins simultaneously but requires injection and has batch-to-batch variability.
Comparison with Non-Peptide Nootropics
Beyond the peptide realm, several other compound classes target overlapping neurotrophic pathways:
- Lion's Mane Mushroom (Hericium erinaceus): Contains hericenones and erinacines that stimulate NGF synthesis. While targeting a different neurotrophin (NGF vs. BDNF), the general strategy of enhancing endogenous neurotrophin production is similar to P21's approach.
- Exercise: Physical activity is the most well-established BDNF-enhancing intervention, with strong evidence from both animal and human studies. Exercise also promotes hippocampal neurogenesis through multiple mechanisms. P21 could theoretically complement exercise-induced neurotrophin signaling.
- Meditation and Sleep: Both have been shown to influence BDNF levels and neuroplasticity. Optimizing these behaviors alongside any nootropic intervention is likely to enhance outcomes.
The Research-to-Application Gap
P21 occupies an unusual position in the nootropic landscape. It has a stronger preclinical evidence base than most nootropic peptides, with published studies in peer-reviewed journals by recognized neuroscience researchers. However, it has zero human data, which places it behind compounds like Semax (limited clinical data from Russia) and Cerebrolysin (multiple clinical trials across several countries).
For individuals interested in evidence-based cognitive enhancement, this creates a genuine dilemma. The animal data for P21 is compelling, but the history of failed translation from animal models to human efficacy in neuroscience is long and sobering. Until human clinical trials are conducted, P21 remains a promising research compound rather than a validated therapeutic.
Those who wish to explore the full range of evidence-based nootropic options can visit our Peptide Hub for comprehensive reviews of compounds at various stages of clinical validation, or take our Free Assessment for personalized recommendations based on individual goals and health status.
Detailed Study-by-Study Analysis of P21 Research
A thorough evaluation of P21's therapeutic potential requires a close examination of each major published study, including its methodology, limitations, and specific findings. This section provides that granular analysis, organized chronologically to trace the evolution of P21 research from its earliest pharmacokinetic characterization through its most recent applications in novel disease models.
Blanchard et al. (2010): Initial Pharmacokinetic Characterization
The foundational pharmacokinetic study of P021 was published by Blanchard and colleagues in Neuroscience Letters in 2010. This study established the basic drug-like properties that made P021 viable as a therapeutic candidate and differentiated it from its parent molecule CNTF.
Study Design and Methods
The researchers conducted a series of in vitro stability assays using artificial gastric juice (pepsin in HCl, pH 1.2) and artificial intestinal fluid (pancreatin in phosphate buffer, pH 6.8) at physiological temperature (37 degrees Celsius). Stability was assessed by high-performance liquid chromatography (HPLC) at multiple time points. In vivo pharmacokinetic studies were performed in mice following subcutaneous injection, with blood samples collected at intervals to determine plasma concentration-time profiles.
Key Findings
The in vitro stability results were striking. P021 maintained over 90% structural integrity after 30 minutes in artificial gastric juice, a condition that degrades most unmodified peptides within minutes. In artificial intestinal fluid, stability exceeded 97% after 2 hours. These values are exceptional for a peptide compound and were directly attributable to the adamantylated glycine modification at the C-terminus and the N-terminal acetylation, which together protect against both endopeptidase and exopeptidase attack.
The in vivo studies revealed a plasma half-life exceeding 3 hours in mice, which is remarkably long for a peptide of this size. For comparison, a typical unmodified tetrapeptide would be expected to have a plasma half-life measured in minutes, not hours. Blood-brain barrier penetration was confirmed by detection of intact P021 in brain tissue following peripheral administration.
Preliminary safety observations in this study showed no acute toxicity at the doses tested, no behavioral changes in treated animals, and no mortality. These initial safety findings provided the foundation for the longer-term efficacy studies that followed.
Limitations and Considerations
This study used a relatively small number of animals and focused on acute pharmacokinetics rather than chronic dosing. The bioavailability was confirmed qualitatively (drug detected in brain) rather than quantified as a precise percentage of administered dose. Detailed tissue distribution data beyond brain detection was not provided. The study did not compare intranasal versus oral versus subcutaneous pharmacokinetics, which would have been valuable for guiding subsequent administration route selection.
Bolognin et al. (2014): Cognitive Aging in Fischer Rats
This study, published in Neurobiology of Aging, was among the first to demonstrate P021's cognitive-enhancing effects in a non-disease model, using aged Fischer 344 rats as a model of normal cognitive aging.
Study Design and Methods
Fischer 344 rats aged 22-24 months (equivalent to approximately 65-70 human years) received P021 chronically through oral administration in their feed at 60 nmol/g. Age-matched young adult rats (4-6 months) served as young controls, and aged rats receiving standard feed served as aged controls. Cognitive function was assessed using the Morris water maze (spatial learning and reference memory) and novel object recognition (recognition memory). Neurogenesis was quantified using BrdU incorporation and NeuN co-labeling in the dentate gyrus. BDNF protein levels were measured by ELISA in hippocampal homogenates. Tau phosphorylation was assessed by Western blotting with phospho-specific antibodies.
Key Findings
Chronic P021 treatment significantly improved spatial learning performance in aged rats compared to untreated aged controls. During acquisition trials in the Morris water maze, P021-treated aged rats showed significantly shorter escape latencies and swim distances, indicating faster learning. During the probe trial (memory retention test), treated aged rats spent significantly more time in the target quadrant compared to untreated aged rats, approaching performance levels of young adults.
At the molecular level, P021 treatment increased BDNF protein levels in the hippocampus of aged rats. This was accompanied by increased numbers of BrdU-positive cells in the dentate gyrus that co-labeled with the mature neuronal marker NeuN, confirming that P021 promoted both proliferation and neuronal differentiation of new cells in the aged brain. Tau hyperphosphorylation at disease-relevant epitopes was also reduced in treated aged rats.
Significance
This study was particularly meaningful because it demonstrated P021's effects in normal aging, rather than in a genetically engineered disease model. Age-related cognitive decline affects essentially all individuals as they get older, and there are currently no approved pharmacological treatments that slow or reverse this decline. P021's ability to rescue age-related cognitive deficits by restoring neurogenesis and BDNF levels in aged rats raised the possibility that the compound could address one of the most common and widespread forms of cognitive impairment.
The study also provided important safety data, as the aged rats tolerated chronic P021 treatment without adverse effects on body weight, general health, or mortality. This was reassuring because aged animals are typically more vulnerable to drug toxicity than younger animals.
Kazim et al. (2014): Disease Modification in 3xTg-AD Mice
Published in Neurobiology of Disease, this study provided the first comprehensive evidence of P021's disease-modifying effects in the triple-transgenic Alzheimer's disease mouse model.
Study Design and Methods
Female 3xTg-AD mice received P021 orally at 60 nmol/g of feed beginning at 9-10 months of age, a time point when extracellular amyloid-beta plaques have begun to deposit but neurofibrillary tangle-like tau pathology has not yet fully developed. This timing was strategically chosen to model a clinically relevant treatment scenario: initiating therapy after early pathological changes are present but before the full disease burden has developed. Treatment continued for 6 months (assessments at 15-16 months) and in some cohorts for 12 months (assessments at 21-22 months). Wild-type mice of matched age and sex served as non-transgenic controls. The behavioral assessment battery included the Morris water maze for spatial learning and memory, the novel object recognition test for recognition memory, and the elevated plus maze for anxiety-like behavior.
Key Findings in Detail
The Morris water maze results were compelling. Untreated 3xTg-AD mice showed severe deficits in spatial learning, requiring significantly more time and distance to locate the hidden platform compared to wild-type controls. P021-treated 3xTg-AD mice performed significantly better than untreated transgenic mice, with escape latencies that approached wild-type levels by the final days of acquisition training. In the probe trial conducted 24 hours after the last training session, P021-treated mice showed significantly greater time spent in the target quadrant and more platform area crossings than untreated transgenic controls.
Novel object recognition testing revealed similar patterns. Untreated 3xTg-AD mice failed to discriminate between novel and familiar objects, spending approximately equal time exploring each, which indicates impaired recognition memory. P021-treated mice showed a significant preference for the novel object, indicating restored recognition memory function.
Biochemical analyses of brain tissue provided mechanistic insight into these cognitive improvements. Western blotting revealed that P021 treatment significantly reduced levels of hyperphosphorylated tau at three distinct epitopes: AT8 (Ser202/Thr205), PHF1 (Ser396/Ser404), and 12E8 (Ser262/Ser356). The magnitude of reduction varied by epitope and time point but was consistently significant compared to vehicle-treated transgenic controls. Total tau protein levels were not significantly affected, confirming that P021 specifically reduced pathological phosphorylation rather than altering tau expression.
The mechanism underlying tau phosphorylation reduction was traced through the BDNF/TrkB/PI3K/Akt/GSK-3 beta pathway. P021-treated mice showed increased BDNF protein levels in the hippocampus, increased phosphorylation (activation) of TrkB at tyrosine residues, increased phosphorylation of Akt at Ser473, and critically, increased inhibitory phosphorylation of GSK-3 beta at Ser9. The coherent activation of this entire signaling cascade provided strong support for the proposed mechanism of action.
Immunohistochemical analysis of amyloid-beta pathology showed reduced plaque burden in the hippocampus and cortex of P021-treated mice. ELISA measurements confirmed reductions in both soluble and insoluble amyloid-beta 40 and amyloid-beta 42 species. While the anti-amyloid effect was secondary to the anti-tau mechanism, it was nonetheless significant and suggested that P021's disease-modifying effects extended to both hallmark pathologies of Alzheimer's disease.
Neurogenesis analysis using Ki-67 (proliferation marker) and doublecortin (immature neuron marker) immunostaining in the dentate gyrus showed that P021 treatment significantly increased both proliferation and neuronal differentiation. Untreated 3xTg-AD mice had markedly reduced neurogenesis compared to wild-type controls, consistent with the known suppression of neurogenesis in Alzheimer's disease models. P021 treatment restored neurogenesis markers to levels approaching those of wild-type animals.
Limitations
The study used only female mice, raising questions about potential sex differences in P021 response. The 3xTg-AD model, while valuable, relies on overexpression of mutant human transgenes and does not perfectly model sporadic (non-familial) Alzheimer's disease, which accounts for over 95% of human cases. The study did not include pharmacokinetic measurements to confirm brain P021 levels, relying instead on the compound's previously demonstrated CNS penetration. Long-term follow-up beyond 12 months of treatment was not reported in this publication.
Kazim and Iqbal (2016): Neurotrophic Factor Small-Molecule Mimetics Review
This comprehensive review article, published in Molecular Neurodegeneration, placed P021 within the broader context of neurotrophic factor mimetics as an emerging therapeutic class for Alzheimer's disease. While not presenting new experimental data, the review provided valuable theoretical framework and identified key challenges in translating preclinical neurotrophic factor research into clinical therapies.
The review emphasized several advantages of the small-molecule mimetic approach over full-length neurotrophic factors: oral bioavailability, blood-brain barrier penetration, resistance to enzymatic degradation, reduced immunogenicity, simpler manufacturing and quality control, and more predictable pharmacokinetics. It also highlighted P021 as one of the most advanced compounds in this class, with the most extensive preclinical dataset demonstrating disease-modifying effects in AD models.
One particularly valuable contribution of this review was its discussion of why anti-amyloid therapies had failed in clinical trials and how neurotrophic approaches might succeed where they had not. The authors argued that Alzheimer's disease involves a complex interplay between multiple pathological processes, including tau hyperphosphorylation, amyloid deposition, neuroinflammation, synaptic loss, and impaired neurogenesis, and that effective therapy must address multiple mechanisms simultaneously. P021's ability to impact tau, amyloid, neurogenesis, and synaptic plasticity through a single upstream mechanism (BDNF upregulation) was presented as a strategic advantage over mono-target approaches.
Baazaoui and Iqbal (2017): Long-Term Preventive Treatment
This study examined the effects of P021 treatment initiated early in life, at 3 months of age, before any detectable pathology in 3xTg-AD mice, and continued for approximately 18 months. This represented the longest P021 treatment study and provided both the strongest efficacy data and the most extensive safety record.
Study Design
3xTg-AD mice and wild-type controls began receiving P021 in their feed (60 nmol/g) at 3 months of age. This early start was designed to test whether P021 could prevent, rather than reverse, Alzheimer-like pathology and cognitive decline. Animals were assessed at multiple time points throughout the 18-month treatment period, with terminal analyses conducted at approximately 21 months of age.
Key Findings
The results were remarkable for their completeness. P021 treatment completely prevented the development of cognitive deficits that normally emerge in 3xTg-AD mice between 6 and 12 months of age. Treated transgenic mice performed comparably to wild-type controls on all cognitive tests throughout the entire treatment period, while untreated transgenic mice showed progressive cognitive decline.
Pathological analysis at the end of the treatment period showed dramatic reductions in both tau and amyloid pathology in P021-treated 3xTg-AD mice compared to untreated transgenic controls. Tau hyperphosphorylation at AT8, PHF1, and 12E8 epitopes was reduced to near-wild-type levels. Amyloid plaque burden was substantially decreased. Neurogenesis markers remained at levels comparable to wild-type animals, in contrast to the severe suppression observed in untreated transgenic mice.
The safety data from this study was particularly valuable. Eighteen months of continuous oral P021 treatment produced no adverse effects on body weight, food intake, general behavior, locomotor activity, anxiety measures, mortality rate, or gross organ pathology. This extended safety record significantly exceeded the duration of typical preclinical drug safety studies and provided strong evidence that chronic P021 exposure is well-tolerated in mice.
Clinical Translation Implications
While preventive treatment starting before pathology onset is not feasible in most Alzheimer's patients (since diagnosis typically occurs after significant pathology has already accumulated), this study is relevant to several clinical scenarios. First, it supports the concept of early intervention in individuals identified as at-risk for Alzheimer's through genetic testing (e.g., APOE4 carriers), biomarker screening (e.g., elevated CSF tau or amyloid PET positivity), or family history. Second, the 18-month safety data provides confidence for designing long-duration clinical trials, which would be necessary for any disease-modifying AD therapy.
Wei et al. (2017): Down Syndrome Model (Ts65Dn Mice)
This study, published in Scientific Reports, expanded P021 research beyond Alzheimer's disease into Down syndrome, a developmental disorder with significant overlap in neuropathology.
Background and Rationale
Down syndrome (DS), caused by trisomy of chromosome 21, is the most common genetic cause of intellectual disability. Individuals with DS have a near-universal risk of developing Alzheimer's disease by age 40-50, due to the triplication of the APP gene located on chromosome 21. The Ts65Dn mouse model carries a partial trisomy of mouse chromosome 16, which includes orthologues of genes on human chromosome 21, and recapitulates many features of DS including cognitive impairment, reduced hippocampal neurogenesis, and age-dependent Alzheimer-like pathological changes.
Study Design
Pregnant Ts65Dn dams received P021 in their feed (60 nmol/g) throughout pregnancy and lactation. Offspring were then continued on P021-supplemented feed through weaning. This early developmental intervention was designed to target the critical period of brain development when DS-related neurogenesis deficits first emerge. Behavioral testing was conducted in adult offspring (several months after the treatment period ended) to determine whether the early intervention had lasting cognitive benefits.
Key Findings
Prenatal-to-early-postnatal P021 treatment rescued developmental delay in Ts65Dn pups. Treated DS pups achieved developmental milestones (eye opening, righting reflex, negative geotaxis) at time points comparable to wild-type littermates, whereas untreated DS pups showed significant delays. More remarkably, the cognitive benefits persisted into adulthood. Adult Ts65Dn mice that had received early P021 treatment showed significantly better performance on hippocampus-dependent memory tasks compared to untreated DS mice, even though the treatment had been discontinued months earlier.
At the molecular level, early P021 treatment increased BDNF expression in the hippocampus and enhanced neurogenesis in the dentate gyrus of Ts65Dn mice. These effects were accompanied by improvements in synaptic marker expression, suggesting that P021's early intervention strengthened the neuronal circuitry during a critical developmental window, with benefits that outlasted the treatment period.
Safety assessments showed no adverse effects of P021 on maternal health, litter size, pup birth weight, or postnatal growth. P021 did not alter body weight, anxiety-like behavior, or general activity levels in the treated Ts65Dn offspring, indicating that its effects were specific to the intended neurodevelopmental targets.
Baazaoui and Iqbal (2020): Prenatal-to-Postnatal Treatment in 3xTg-AD Mice
Published in Alzheimer's Research and Therapy, this study extended the developmental treatment paradigm to the 3xTg-AD mouse model, testing whether early-life P021 treatment could prevent the later emergence of Alzheimer-like pathology and cognitive deficits.
Study Design
P021 was administered through the maternal diet during pregnancy and continued through the postnatal period. Offspring were then followed into adulthood and assessed for cognitive function, brain pathology, and molecular markers of neurogenesis and synaptic integrity.
Key Findings
Early-life P021 treatment significantly delayed or prevented the development of Alzheimer-like cognitive deficits, tau hyperphosphorylation, and amyloid pathology in 3xTg-AD mice. The treatment produced lasting increases in BDNF expression and neurogenesis that persisted well beyond the treatment period. These findings complemented the Down syndrome study by demonstrating that early neurotrophic intervention during critical developmental periods can produce enduring neuroprotective effects in genetic models of neurodegeneration.
The study also provided additional long-term safety data, confirming that perinatal P021 exposure did not produce any delayed adverse effects in offspring monitored for months after treatment cessation. This was an important finding given concerns about the long-term consequences of manipulating neurotrophic signaling during brain development.
Galvani et al. (2024): CDKL5 Deficiency Disorder
The most recent expansion of P021 research into a new disease model was published by Galvani and colleagues in the Journal of Neurodevelopmental Disorders in 2024. This study examined P021's effects in CDKL5 deficiency disorder (CDD), a rare neurodevelopmental condition caused by loss-of-function mutations in the CDKL5 gene.
Background
CDD is characterized by early-onset seizures, intellectual disability, motor impairment, and behavioral features that overlap with autism spectrum disorder. The CDKL5 gene encodes a serine-threonine kinase expressed in neurons that interacts with several signaling pathways relevant to neuronal development and plasticity, including those involving BDNF. Reduced BDNF expression and impaired neurogenesis have been documented in Cdkl5 knockout mouse models, providing rationale for testing P021's BDNF-enhancing mechanism in this disorder.
Study Design
The researchers employed both in vitro and in vivo approaches. In vitro studies used human neurons derived from induced pluripotent stem cells (iPSCs) carrying CDKL5 mutations. In vivo studies used Cdkl5 knockout mice. P021 was tested for its ability to increase BDNF expression, enhance neurogenesis, and improve phenotypic outcomes in both model systems.
Key Findings
P021 increased BDNF expression in both the human iPSC-derived neurons and the Cdkl5 knockout mice, confirming that its BDNF-enhancing mechanism translates across species and disease models. In the knockout mice, P021 treatment enhanced hippocampal neurogenesis as measured by increased numbers of Ki-67-positive and DCX-positive cells in the dentate gyrus.
However, the therapeutic effects were incomplete. While some measures of neuronal function and behavioral phenotype showed improvement with P021 treatment, others did not reach statistical significance. This partial efficacy may reflect the fact that CDD pathophysiology involves mechanisms beyond BDNF deficiency, including disruption of CDKL5-dependent phosphorylation cascades that P021 does not directly address. The study's importance lies in expanding the known spectrum of P021's activity and identifying both its potential and its limitations in different neurological conditions.
Iqbal et al. (2022): Comprehensive Review of P021 as an AD Therapeutic
Published in Biomolecules, this review by the P021 development team provided the most comprehensive and up-to-date summary of the compound's preclinical development. The article covered the rationale for neurotrophic factor mimetics, detailed P021's mechanism of action, summarized all published efficacy and safety data, and discussed the path toward clinical translation.
A particularly valuable aspect of this review was its discussion of the "synaptic compensation" hypothesis. The authors proposed that Alzheimer's disease progresses through an early phase of synaptic compensation, during which the brain upregulates synaptic proteins and neuroplasticity mechanisms to compensate for initial pathological insults, followed by a decompensation phase when these compensatory mechanisms fail. P021's greatest therapeutic potential, they argued, lies in extending and strengthening the synaptic compensation phase, thereby delaying or preventing the transition to decompensation and clinical dementia.
This review also provided the most detailed discussion to date of P021's commercial development through Phanes Biotech, noting that the compound is being advanced toward IND-enabling studies with the goal of initiating human clinical trials for Alzheimer's disease.
Understanding BDNF: The Central Mediator of P21's Effects
Brain-derived neurotrophic factor (BDNF) is the most abundant neurotrophin in the adult brain and serves as the primary downstream mediator of P21's therapeutic effects. A thorough understanding of BDNF biology is essential for evaluating P21's potential and limitations as a therapeutic compound.
BDNF Synthesis, Processing, and Secretion
BDNF is encoded by a complex gene that in humans spans over 70 kilobases and contains multiple promoters, 11 exons, and alternative splice variants. This genomic complexity allows for tissue-specific and activity-dependent regulation of BDNF expression. The BDNF gene produces a precursor protein (proBDNF) of approximately 32 kDa, which is proteolytically cleaved to generate the mature BDNF protein (approximately 14 kDa). Both proBDNF and mature BDNF are biologically active, but they signal through different receptors and have opposing effects: mature BDNF promotes neuronal survival and synaptic strengthening through TrkB, while proBDNF promotes apoptosis and synaptic weakening through the p75 neurotrophin receptor (p75NTR).
BDNF is synthesized primarily in neurons and is secreted through both constitutive and regulated (activity-dependent) secretory pathways. Activity-dependent BDNF release is triggered by neuronal depolarization and calcium influx and is particularly important for synaptic plasticity. This activity-dependent regulation means that BDNF release is spatially and temporally targeted to active synapses, providing local trophic support to synaptic connections that are being strengthened through use. This contrasts with the indiscriminate activation that would result from administering exogenous BDNF, which cannot replicate the spatial and temporal precision of endogenous activity-dependent release.
P21's approach of increasing endogenous BDNF expression (by removing LIF-mediated transcriptional suppression) preserves this activity-dependent regulation, since the newly synthesized BDNF is still packaged into regulated secretory vesicles and released in response to neuronal activity. This is a theoretical advantage over direct BDNF administration or direct TrkB receptor agonists, which bypass the physiological regulatory mechanisms that ensure BDNF acts at the right place and time.
BDNF and Long-Term Potentiation
Long-term potentiation (LTP) is the cellular mechanism believed to underlie memory formation. It involves a persistent strengthening of synaptic transmission following high-frequency stimulation. BDNF plays a critical role in several phases of LTP:
- Early-phase LTP (E-LTP): BDNF modulates NMDA receptor function and AMPA receptor trafficking to the synaptic membrane, enhancing excitatory postsynaptic potentials. This phase occurs within minutes and does not require new protein synthesis.
- Late-phase LTP (L-LTP): BDNF activates transcription factor CREB, which drives expression of genes necessary for structural synaptic modifications, including new dendritic spine formation and synapse stabilization. This phase develops over hours and requires gene transcription and protein synthesis.
- Synaptic tagging and capture: BDNF contributes to the synaptic tagging process that allows specific synapses to capture newly synthesized plasticity-related proteins, ensuring that LTP is expressed at the appropriate synapses.
By increasing BDNF expression, P21 would be expected to enhance all phases of LTP, which would translate to improved memory encoding and consolidation. This is consistent with the cognitive improvements observed in P021-treated animals across multiple behavioral paradigms testing different types of memory (spatial, recognition, contextual).
BDNF Decline in Aging and Disease
BDNF levels in the brain decline progressively with aging, and this decline is accelerated in neurodegenerative diseases. In Alzheimer's disease specifically, BDNF protein levels are reduced by approximately 30-40% in the hippocampus and cortex compared to age-matched controls. Similar reductions are observed in the cerebrospinal fluid (CSF) and serum of AD patients, and reduced serum BDNF levels have been proposed as a peripheral biomarker for early AD detection.
The causes of age-related BDNF decline are multiple and interacting: increased inflammatory signaling (which suppresses BDNF transcription), reduced physical activity (a potent natural BDNF stimulus), accumulated oxidative damage to BDNF gene regulatory regions, epigenetic changes (particularly increased methylation of BDNF promoters), and altered neurotransmitter signaling (particularly reduced glutamatergic and cholinergic activity). P021 addresses one specific mechanism of BDNF suppression, namely the LIF-mediated transcriptional inhibition through JAK-STAT3 signaling. While this does not address all causes of age-related BDNF decline, it targets a mechanism that appears to be tonically active and amenable to pharmacological modulation.
The Val66Met BDNF Polymorphism and P21 Response
An important consideration for the potential clinical use of P021 is the common Val66Met polymorphism (rs6265) in the human BDNF gene. This single nucleotide polymorphism, present in approximately 20-30% of the general population (with higher frequencies in Asian populations), affects the intracellular trafficking and activity-dependent secretion of BDNF. Met-allele carriers show reduced BDNF secretion, smaller hippocampal volume, and impaired hippocampus-dependent memory compared to Val/Val homozygotes.
The Val66Met polymorphism could theoretically affect P021 response in two opposing ways. On one hand, Met carriers might benefit more from P021 treatment because they have lower baseline BDNF signaling and therefore more room for improvement. On the other hand, the Met variant impairs BDNF secretion at the protein trafficking level, downstream of the transcriptional enhancement that P021 provides, potentially limiting the functional impact of increased BDNF mRNA. This question has not been addressed in any published P021 study, as the research has been conducted exclusively in mice that do not carry this human polymorphism. However, it will be an important consideration for clinical trial design and may influence patient selection and stratification.
BDNF and Exercise: A Natural Comparator
Physical exercise is the most well-established natural intervention for increasing brain BDNF levels in humans. Aerobic exercise in particular has been shown to increase both circulating BDNF levels and hippocampal volume in controlled clinical trials. The magnitude of BDNF increase with regular exercise (approximately 10-30% above sedentary baseline) provides a useful benchmark against which P021's BDNF-enhancing effects can be considered.
In the animal studies, P021 treatment produced BDNF increases that were generally in the same range as or somewhat larger than those achieved with exercise paradigms, though direct head-to-head comparisons have not been published. The practical implication is that P021 might provide BDNF-enhancing benefits comparable to exercise for individuals who are unable to exercise due to physical disability, severe illness, or other limitations. For individuals who can exercise, P021 and exercise likely have additive effects since they increase BDNF through different mechanisms (exercise primarily through activity-dependent release; P021 through transcriptional upregulation via LIF inhibition).
This relationship between P021 and exercise-induced neuroplasticity is discussed further in our Biohacking Hub, which covers the intersection of pharmacological and lifestyle approaches to cognitive optimization.
GSK-3 Beta and Tau Biology: P21's Anti-Alzheimer's Mechanism
The inhibition of glycogen synthase kinase-3 beta (GSK-3 beta) through the BDNF/TrkB/PI3K/Akt pathway is P21's primary mechanism for reducing tau hyperphosphorylation, one of the two hallmark pathologies of Alzheimer's disease. Understanding the biology of GSK-3 beta and tau is essential for evaluating P21's potential as a disease-modifying therapy.
GSK-3 Beta: A Master Kinase
GSK-3 beta is a serine/threonine kinase that was originally identified for its role in glycogen metabolism but has since been recognized as a central regulatory kinase involved in diverse cellular processes, including cell survival, proliferation, differentiation, migration, glucose regulation, gene transcription, and circadian rhythm. In the brain, GSK-3 beta is particularly abundant in neurons, where it regulates synaptic plasticity, neurogenesis, and neuroinflammation.
GSK-3 beta is constitutively active in resting cells, meaning it is "always on" unless specifically inhibited. Inhibitory regulation occurs primarily through phosphorylation at serine 9 by upstream kinases, including Akt (the pathway activated by P021), protein kinase A (PKA), and protein kinase C (PKC). When Ser9 is phosphorylated, the N-terminal domain of GSK-3 beta acts as a pseudosubstrate, occupying the substrate binding groove and preventing the kinase from accessing its true substrates.
This regulatory mechanism is the linchpin of P021's anti-tau effects. By increasing BDNF expression and activating the TrkB/PI3K/Akt cascade, P021 enhances Akt-mediated phosphorylation of GSK-3 beta at Ser9, reducing the kinase's activity and consequently decreasing the phosphorylation of its substrates, including tau protein.
Tau Protein: From Normal Function to Pathological Aggregation
Tau is a microtubule-associated protein that normally functions to stabilize microtubules in neuronal axons, facilitating axonal transport of organelles, vesicles, and signaling molecules. In its normal (non-hyperphosphorylated) state, tau binds to microtubules and promotes their assembly and stability. The protein contains 85 potential serine and threonine phosphorylation sites, and the degree of phosphorylation regulates its affinity for microtubules.
In Alzheimer's disease, tau becomes hyperphosphorylated at multiple sites, meaning it accumulates far more phosphate groups than normal tau. This hyperphosphorylation reduces tau's affinity for microtubules, causing it to detach and accumulate in the cytoplasm. Unbound hyperphosphorylated tau then aggregates first into oligomers, then into paired helical filaments (PHFs), and ultimately into neurofibrillary tangles (NFTs), which are one of the defining pathological features of Alzheimer's disease and are more closely correlated with cognitive decline than amyloid plaques.
The discovery that tau in Alzheimer's neurofibrillary tangles is abnormally hyperphosphorylated was made by Drs. Khalid Iqbal and Inge Grundke-Iqbal in 1986, the same research team that later developed P021. This personal connection between the discovery of tau hyperphosphorylation and the development of a therapeutic compound targeting that pathway adds a compelling narrative arc to P021's development history.
GSK-3 Beta as a Tau Kinase
Among the approximately 20 protein kinases that can phosphorylate tau in vitro, GSK-3 beta is considered the most physiologically relevant tau kinase for several reasons. First, GSK-3 beta phosphorylates tau at the majority of the sites that are hyperphosphorylated in Alzheimer's disease, including Thr181, Ser199, Ser202, Thr205, Thr231, Ser235, Ser396, Ser400, Ser404, and Ser413. Second, GSK-3 beta is colocalized with pretangle and tangle neurons in AD brains. Third, genetic manipulation of GSK-3 beta activity in transgenic mice recapitulates tau pathology: overexpression of GSK-3 beta increases tau phosphorylation, while GSK-3 beta knockout or inhibition reduces it.
P021's indirect inhibition of GSK-3 beta through the BDNF/Akt pathway offers several potential advantages over direct GSK-3 beta inhibitors. Direct inhibitors (such as lithium, tideglusib, and CHIR99021) act on all GSK-3 beta in the body, potentially disrupting its many non-tau functions, including insulin signaling, glycogen metabolism, Wnt pathway regulation, and cell cycle control. P021's indirect approach, working through BDNF-mediated Akt activation, modulates GSK-3 beta activity in a more physiological manner, enhancing an endogenous regulatory mechanism rather than directly blocking the enzyme's catalytic activity.
The Dual Pathology Problem: Tau and Amyloid
A longstanding debate in Alzheimer's research concerns the relative importance of amyloid-beta plaques versus tau neurofibrillary tangles in driving disease progression. The "amyloid cascade hypothesis" proposes that amyloid-beta accumulation is the initiating event, with tau pathology developing as a downstream consequence. An alternative view places tau pathology as the primary driver of neurodegeneration, with amyloid-beta playing a facilitating but not essential role.
P021's effects on both pathologies provide evidence for their interconnection. In 3xTg-AD mice, P021 treatment reduced both tau hyperphosphorylation and amyloid-beta pathology, suggesting that these two processes are linked through shared signaling pathways. GSK-3 beta, the kinase targeted indirectly by P021, has been shown to influence both tau phosphorylation and amyloid-beta production. Active GSK-3 beta promotes amyloidogenic processing of APP (amyloid precursor protein) through phosphorylation-dependent regulation of gamma-secretase complex components. By inhibiting GSK-3 beta, P021 may therefore reduce both amyloidogenic processing and tau hyperphosphorylation through a common mechanism.
This dual-pathology effect distinguishes P021 from most other AD drug candidates, which typically target either amyloid or tau but not both. The recent clinical success of anti-amyloid antibodies (lecanemab, donanemab) in slowing AD progression has validated the amyloid target but has also highlighted the modest clinical benefit achieved by addressing amyloid alone. A compound like P021 that addresses both pathologies simultaneously might theoretically provide greater clinical benefit, though this hypothesis requires human testing to evaluate.
Practical Considerations for Researchers and Clinicians
While P21 remains a preclinical research compound without human clinical data, researchers investigating its properties and clinicians monitoring patients who have chosen to self-administer the compound can benefit from practical guidance based on the available evidence.
Research Applications
P021 has several characteristics that make it useful as a research tool beyond its potential therapeutic applications:
- Neurogenesis probe: P021's well-characterized mechanism (LIF inhibition leading to BDNF upregulation and neurogenesis) makes it a useful pharmacological probe for studying adult neurogenesis in vivo. Researchers can use P021 to selectively enhance neurogenesis and study the functional consequences of new neuron addition to hippocampal circuits.
- BDNF pathway activation: P021 provides a convenient way to upregulate endogenous BDNF expression in vivo without the complications of viral vector-mediated BDNF overexpression or direct BDNF infusion. This is valuable for studying BDNF-dependent processes in naturalistic experimental conditions.
- GSK-3 beta inhibition studies: P021 offers an indirect, physiologically relevant approach to GSK-3 beta inhibition through the BDNF/Akt pathway, which may better model therapeutic GSK-3 beta modulation than direct kinase inhibitors.
- Disease model treatment: P021's oral bioavailability and long-term safety profile make it practical for chronic treatment studies in disease models where sustained neurotrophic support is the experimental goal.
Reconstitution and Storage
P021 is typically supplied as a lyophilized (freeze-dried) powder. For research applications, the following handling guidelines are based on general peptide chemistry principles and limited available documentation:
- Storage of lyophilized powder: Store at -20 degrees Celsius or below in a sealed container with desiccant. Lyophilized P021 is stable for extended periods (months to years) under these conditions.
- Reconstitution: Reconstitute with sterile water, bacteriostatic water, or sterile saline. Gently swirl or roll the vial rather than vortexing vigorously, as aggressive mixing can damage peptide structure. Allow the peptide to dissolve completely before use.
- Storage of reconstituted solution: Store at 2-8 degrees Celsius (standard refrigeration). Use within 2-4 weeks for optimal stability, though the adamantane modification provides better stability than most reconstituted peptides.
- Avoid freeze-thaw cycles: If longer storage is needed, aliquot the reconstituted solution into single-use volumes and freeze. Avoid repeated freeze-thaw cycles, which can promote peptide aggregation and degradation.
Biomarker Monitoring Considerations
For researchers or clinicians monitoring individuals using P021, several biomarkers could theoretically be informative, though none have been validated specifically for P021 response assessment:
- Serum BDNF: Peripheral BDNF levels correlate roughly with central BDNF levels and could serve as a proxy marker for P021's pharmacodynamic activity. However, serum BDNF is influenced by many factors (exercise, diet, sleep, stress, platelet count) and has significant intra-individual variability, limiting its utility as a stand-alone marker.
- Cognitive testing: Standardized cognitive assessments (MoCA, MMSE, computerized cognitive batteries) could track cognitive changes over time. Given P021's slow-onset neurogenic mechanism, assessments should be conducted at baseline and then at intervals of at least 4-8 weeks.
- Neuroimaging: Hippocampal volume on MRI could theoretically reflect P021-induced neurogenesis, but the changes would likely be too small to detect with standard clinical MRI protocols. Research-grade volumetric analysis might detect subtle changes with sufficient sample size and follow-up duration.
- CSF biomarkers: In clinical trials, CSF phospho-tau (p-tau181, p-tau217, p-tau231) and total tau could serve as pharmacodynamic markers for P021's anti-tau effects. CSF amyloid-beta 42/40 ratio could assess anti-amyloid effects. These invasive measurements would be appropriate for clinical research but not for routine monitoring.
Who Should Not Use P21
While P021 has shown a clean safety profile in animal studies, the following populations should avoid P021 pending human safety data:
- Pregnant or breastfeeding individuals: P021's effects on human fetal development are unknown. While early postnatal treatment was beneficial in the Ts65Dn mouse model, this cannot be extrapolated to human pregnancy without clinical data.
- Individuals with active cancer: BDNF and its receptor TrkB are expressed in various cancer types, and BDNF/TrkB signaling can promote tumor cell survival and proliferation. While P021 did not cause tumors in 18-month mouse studies, individuals with existing malignancies should avoid compounds that enhance neurotrophic signaling.
- Individuals with epilepsy: BDNF has been implicated in both pro-epileptogenic and anti-epileptogenic processes. Elevated BDNF levels in the hippocampus could theoretically lower seizure threshold in susceptible individuals. This concern is theoretical but warrants caution.
- Individuals on lithium therapy: Both lithium and P021 (indirectly) inhibit GSK-3 beta. Combining them could produce excessive GSK-3 beta inhibition with unpredictable consequences.
- Children and adolescents: The effects of P021 on the developing human brain are unknown. While postnatal treatment was beneficial in mouse models of Down syndrome, human brain development differs substantially from rodent brain development in timing, duration, and molecular mechanisms.
Regulatory and Legal Status
P021's regulatory status varies by jurisdiction but is generally classified as a research chemical or research peptide. Key points include:
- P021 is not approved by the FDA, EMA, or any other regulatory authority for human use
- It is not classified as a controlled substance in any jurisdiction (as of 2026)
- It is sold by research chemical suppliers with labels indicating "for research use only" or "not for human consumption"
- Off-label human use exists but is not endorsed by any medical organization
- No Investigational New Drug (IND) application has been publicly announced, though Phanes Biotech is reportedly working toward this milestone
- Practitioners prescribing or compounding P021 for human use should be aware of the complete absence of human safety and efficacy data
Quality Control Concerns
As with all research peptides obtained from commercial suppliers, quality control is a significant concern for P021. The peptide market includes both legitimate research suppliers producing high-purity compounds and less scrupulous sellers offering products of unknown quality. Key quality considerations include:
- Purity: Look for suppliers providing certificates of analysis (COAs) with HPLC purity data showing at least 95% purity, preferably 98% or higher.
- Identity confirmation: Mass spectrometry data confirming the correct molecular weight (578.3 Da) should be provided on the COA.
- Endotoxin testing: For any peptide intended for injection (even in research settings), endotoxin levels should be below 0.5 EU/mg.
- Third-party testing: Independent testing by a third-party laboratory provides the strongest assurance of quality. Some suppliers submit their products for third-party verification; others do not.
- Proper storage during shipping: P021 should be shipped cold (ice packs or dry ice) to maintain stability during transit. Room-temperature shipping may compromise the peptide, particularly during summer months.
For access to P21 that meets stringent quality standards, FormBlends maintains rigorous testing protocols and provides full certificates of analysis with every order.
Compounding Pharmacy Considerations
Some individuals obtain P21 through compounding pharmacies rather than research peptide suppliers. Compounding pharmacies operate under different regulatory frameworks than research chemical companies and may offer certain advantages, including pharmaceutical-grade purity standards, proper sterility assurance for injectable preparations, professional guidance on reconstitution and administration, and the ability to customize formulations (e.g., nasal sprays with appropriate preservatives and pH buffering). However, compounding pharmacies typically charge significantly more than research suppliers, and not all compounding pharmacies are willing to prepare P21 given its status as an unapproved research compound. Individuals considering this route should ensure that their compounding pharmacy has experience with peptide formulations and follows United States Pharmacopeia (USP) Chapter 797 standards for sterile compounding or equivalent guidelines in their jurisdiction.
Adult Neurogenesis: The Biological Context for P21's Effects
P21's primary pharmacological action is the promotion of adult hippocampal neurogenesis. To fully appreciate the significance of this effect, it helps to understand the biology of adult neurogenesis in detail, including the process itself, its regulation, its functional consequences, and the ongoing scientific debates about its extent in the human brain.
Discovery and Historical Context
For most of the twentieth century, the dogma in neuroscience held that the adult brain could not produce new neurons. This view, articulated most famously by Santiago Ramon y Cajal, dominated thinking about brain plasticity and had profound implications for how neurological diseases were understood and treated. If the brain could not regenerate neurons, then neuronal loss from aging, injury, or disease was permanent and irreversible.
This dogma began to crumble in the 1960s when Joseph Altman and colleagues used autoradiographic techniques to demonstrate incorporation of tritiated thymidine (a marker of DNA synthesis and cell division) into cells in the rat hippocampus and olfactory bulb that had morphological characteristics of neurons. However, these findings were largely dismissed or ignored by the broader neuroscience community for decades.
The modern era of adult neurogenesis research began in the 1990s with the development of new techniques, particularly BrdU labeling combined with cell-type-specific markers, that provided more convincing evidence of new neuron production in adult rodent brains. Fred Gage, Gerd Kempermann, and colleagues at the Salk Institute were instrumental in establishing that the hippocampal dentate gyrus of adult rats and mice contains a population of neural stem cells that continuously produces new neurons throughout life. The landmark 1998 study by Eriksson et al. extended these findings to the adult human brain, demonstrating BrdU incorporation into cells expressing the neuronal marker NeuN in the dentate gyrus of cancer patients who had received BrdU injections as part of their clinical treatment.
The Neurogenic Niche: Where New Neurons Are Born
Adult neurogenesis in the hippocampus occurs within a specialized microenvironment called the neurogenic niche, located in the subgranular zone (SGZ) of the dentate gyrus. This niche consists of several cell types and molecular signals that together regulate the proliferation, differentiation, survival, and integration of new neurons.
Neural stem cells (Type 1 cells): These are radial glia-like cells that express glial fibrillary acidic protein (GFAP), nestin, and Sox2. They are relatively quiescent, dividing infrequently, and serve as the reservoir from which new neurons are ultimately derived. The balance between quiescence and activation in these cells is critical for maintaining the stem cell pool while producing new neurons, and it is this balance that P21 modulates through LIF signaling inhibition.
Transit-amplifying progenitors (Type 2 cells): When neural stem cells divide, they produce rapidly dividing transit-amplifying progenitors that expand the population of cells available for neuronal differentiation. These cells express markers like Mash1 and gradually transition from expressing glial markers to neuronal markers as they progress through division.
Neuroblasts (Type 3 cells): These immature neurons express doublecortin (DCX) and PSA-NCAM and have begun to extend processes (dendrites and axons) into the surrounding tissue. They migrate a short distance from the SGZ into the granule cell layer (GCL) of the dentate gyrus. This is the stage at which most new cells die; estimates suggest that only 20-30% of newly born cells survive to become mature neurons, while the rest undergo apoptosis within the first few weeks of life.
Mature granule neurons: Over a period of approximately 4-8 weeks (in rodents), surviving neuroblasts extend dendrites into the molecular layer, send axons along the mossy fiber pathway to CA3, and form functional synaptic connections with existing hippocampal circuits. They express mature neuronal markers including NeuN and calbindin, and they become electrophysiologically integrated, exhibiting normal action potential firing patterns and synaptic responses.
P21's effect on neurogenesis has been documented at multiple stages of this process. It increases the number of Ki-67-positive cells (indicating enhanced proliferation of progenitor cells), DCX-positive cells (indicating more immature neurons surviving through the critical differentiation period), and BrdU/NeuN double-positive cells (indicating more cells successfully completing the maturation process to become functional neurons). This multi-stage enhancement suggests that P21 does not simply increase cell division but also promotes the survival and maturation of newly born neurons, likely through its BDNF-mediated trophic support.
Functional Significance of Adult Neurogenesis
What do new neurons actually do once they integrate into hippocampal circuits? This question has been the subject of intensive research, and while the full picture is still emerging, several functions have been associated with adult-born neurons:
Pattern separation: The dentate gyrus is believed to perform a computational function called pattern separation, which involves creating distinct neural representations for similar but not identical experiences. This function is critical for distinguishing between similar memories, such as remembering where you parked your car today versus yesterday. Adult-born neurons, with their unique electrophysiological properties during the early maturation period (lower threshold for LTP, broader tuning for spatial inputs), appear to play a particularly important role in pattern separation. Studies using optogenetic silencing of adult-born neurons have shown impaired pattern separation with preserved pattern completion, confirming a specific functional contribution.
Spatial memory: Multiple studies have shown that ablation of neurogenesis (through genetic manipulation, irradiation, or anti-mitotic drugs) impairs spatial memory tasks that depend on the hippocampus, such as the Morris water maze. Conversely, enhancement of neurogenesis (through exercise, enriched environment, or pharmacological stimulation) improves spatial memory. P21's improvement of Morris water maze performance in multiple animal models is consistent with this relationship.
Contextual learning: Adult-born neurons contribute to the hippocampus's ability to associate events with their spatial and temporal context. This includes contextual fear conditioning, where an animal learns to associate a specific environment with an aversive stimulus, and contextual discrimination, where an animal must distinguish between contexts associated with different outcomes.
Mood regulation: Hippocampal neurogenesis has been linked to mood regulation and stress resilience. Reduced neurogenesis is associated with depression-like behavior in animal models, and antidepressant drugs that are effective in humans (SSRIs, SNRIs, exercise) also promote hippocampal neurogenesis. While the causal direction of this relationship is debated, the association is strong and consistent across many studies and species.
Forgetting and memory clearance: Paradoxically, neurogenesis has also been implicated in the active forgetting of old memories. New neurons integrate into existing circuits and, in doing so, remodel the synaptic connections that stored prior memories. This "neurogenesis-dependent forgetting" may serve an adaptive function by clearing outdated information and making room for new learning. This process could be relevant to P21's therapeutic potential in conditions where excessive or intrusive memories are problematic, such as post-traumatic stress disorder (PTSD).
The Human Neurogenesis Debate
One of the most contentious questions in modern neuroscience is the extent to which adult neurogenesis occurs in the human brain. While the 1998 Eriksson study provided evidence for neurogenesis in adult humans, subsequent studies have produced conflicting results, and the debate remains unresolved.
In 2018, Sorrells et al. published a study in Nature reporting that they found no evidence of neurogenesis in the dentate gyrus of adult humans based on immunohistochemical analysis of postmortem brain tissue. They concluded that if neurogenesis occurs in the adult human hippocampus, it is extremely rare. This finding was challenged the same year by Boldrini et al., who reported in Cell Stem Cell that they found evidence of continued neurogenesis into the eighth decade of life in human postmortem hippocampus, with thousands of immature neurons (DCX-positive cells) present even in elderly individuals.
The discrepancy between these studies may reflect methodological differences in tissue fixation, post-mortem interval, antibody selection, and quantification methods. More recent studies using alternative approaches, including single-nucleus RNA sequencing and carbon-14 birth dating of neurons, have provided additional evidence for continued, though perhaps reduced, neurogenesis in the adult human hippocampus.
This debate has direct implications for P21's potential in humans. If adult hippocampal neurogenesis is substantial in humans, then P21's neurogenic effects observed in mice could translate meaningfully to human cognitive enhancement. If human neurogenesis is minimal or absent, P21's cognitive benefits (if any) in humans would need to rely on its other mechanisms, principally BDNF-mediated synaptic plasticity enhancement, rather than new neuron production. Even in the absence of neurogenesis, P21's effects on BDNF, synaptic proteins, dendritic morphology, and tau phosphorylation would still be therapeutically relevant if they translate from mouse to human brain.
Regulators of Adult Neurogenesis
Adult neurogenesis is regulated by a complex interplay of intrinsic genetic programs and extrinsic environmental and molecular signals. Understanding these regulators provides context for P21's mechanism and helps identify potential complementary interactions with other interventions.
Positive regulators (enhance neurogenesis):
- Physical exercise (particularly aerobic/running), the most potent natural neurogenesis enhancer
- Environmental enrichment (novel stimuli, social interaction, cognitive challenge)
- BDNF signaling (P21's primary pathway)
- Serotonin (mechanism of antidepressant-induced neurogenesis)
- Wnt/beta-catenin signaling
- Notch signaling (regulates stem cell maintenance)
- IGF-1 (insulin-like growth factor 1)
- VEGF (vascular endothelial growth factor)
- Caloric restriction and intermittent fasting
- Adequate sleep (particularly slow-wave sleep)
Negative regulators (suppress neurogenesis):
- Chronic stress and elevated glucocorticoids
- Neuroinflammation and elevated pro-inflammatory cytokines
- Aging (progressive decline in neurogenesis rate)
- LIF signaling (the specific target of P21)
- High-fat diet and metabolic syndrome
- Chronic alcohol consumption
- Sleep deprivation
- Social isolation
- Certain medications (some anti-inflammatory drugs, opioids)
P21's mechanism targets one specific negative regulator (LIF signaling) and enhances one specific positive regulator (BDNF). This focused mechanism has the advantage of selectivity but also means that P21 does not address all the factors that suppress neurogenesis. For optimal neurogenic stimulation, P21 would theoretically be most effective when combined with lifestyle interventions that address other regulators, particularly exercise, stress management, adequate sleep, and cognitive enrichment. This integrated approach to cognitive optimization is explored in detail at our Biohacking Hub.
Clinical Translation Challenges: From Mouse to Human
The path from promising preclinical compound to approved human therapeutic is long, expensive, and fraught with failure. Understanding the specific challenges facing P21's clinical translation helps set realistic expectations about the compound's future and provides context for why promising animal data does not guarantee human efficacy.
The Alzheimer's Drug Development Graveyard
Between 2002 and 2022, over 200 experimental compounds for Alzheimer's disease entered clinical trials. The failure rate exceeded 99%. For compounds targeting disease modification (as opposed to symptomatic relief), the failure rate was even higher. This extraordinary attrition rate reflects fundamental challenges in AD drug development that apply to P21 as much as to any other candidate.
Model validity: The 3xTg-AD mouse model, while among the most widely used AD models, has significant limitations. It uses overexpression of mutant human genes (APP, tau, PS1) to drive pathology, which may not accurately model the sporadic AD that affects over 95% of human patients. The mice do not develop the same pattern of neuronal loss seen in human AD. Their disease develops over months rather than decades, and their cognitive decline, while measurable on rodent behavioral tests, cannot capture the complexity of human dementia. P21 has demonstrated clear efficacy in this model, but many compounds that showed equal or greater efficacy in the same model failed in human trials.
Timing of intervention: In the animal studies, P21 treatment was initiated either before pathology onset (3 months) or during an intermediate stage (9-10 months). In human clinical practice, Alzheimer's diagnosis typically occurs after years or decades of pathological progression, when substantial neuronal loss and brain atrophy have already occurred. Whether P21 can provide meaningful benefit at these later disease stages is unknown.
Species differences in drug metabolism: P21's pharmacokinetic properties have been characterized in mice but not in humans. Drug metabolism, distribution, and elimination can differ substantially between species due to differences in enzyme expression, protein binding, organ perfusion, and renal function. The oral bioavailability, plasma half-life, and brain penetration observed in mice may not be replicated in humans.
Human neurogenesis uncertainty: As discussed, the extent of adult neurogenesis in the human brain remains debated. If human hippocampal neurogenesis is substantially less active than rodent neurogenesis, one of P21's primary mechanisms of action may be less relevant in humans.
Regulatory Requirements
Before P21 can enter human clinical trials, it must complete a comprehensive package of IND-enabling (Investigational New Drug) studies that satisfy FDA requirements. These typically include:
Good Manufacturing Practice (GMP) synthesis: P21 must be synthesized under GMP conditions that ensure batch-to-batch consistency, purity (typically >98%), and freedom from endotoxins, heavy metals, and other contaminants. GMP synthesis for peptides is expensive and requires specialized facilities.
Formal toxicology studies: While P21's safety in mice is well-documented, the FDA requires toxicology studies in at least two species (typically one rodent and one non-rodent, such as dog or non-human primate) conducted under Good Laboratory Practice (GLP) conditions. These studies must include acute toxicity, repeat-dose toxicity (typically 28-day and 90-day), genotoxicity, reproductive toxicity, and carcinogenicity assessments.
Pharmacokinetic characterization: Detailed PK studies including absorption, distribution, metabolism, and excretion (ADME) in multiple species, plasma protein binding, metabolite identification, and assessment of drug-drug interaction potential through cytochrome P450 enzyme inhibition/induction studies.
Formulation development: The final drug product must be formulated for the intended route of administration (oral, intranasal, subcutaneous) with appropriate excipients, in dosage forms suitable for human use, with documented stability under defined storage conditions.
Clinical Trial Design Considerations
If P21 reaches clinical trials, several design challenges must be addressed:
Phase I (Safety): First-in-human studies would establish safe dose ranges, characterize pharmacokinetics in healthy volunteers, and identify dose-limiting toxicities. Given P21's excellent animal safety profile, this phase might proceed relatively smoothly, but unexpected human-specific toxicities are always possible.
Phase II (Efficacy): Proof-of-concept efficacy studies in AD patients would need to demonstrate measurable cognitive or biomarker improvements over 6-18 months. Patient selection (early vs. moderate AD), outcome measures (cognitive scales, biomarkers, neuroimaging), and trial duration are all critical design decisions. P21's slow-onset neurogenic mechanism may require longer treatment periods than the 12-18 months typical of AD Phase II trials.
Phase III (Confirmatory): Large-scale confirmatory trials with thousands of patients over 18-36 months would be required for regulatory approval. These trials are extremely expensive (typically $100-500 million per trial for AD) and represent the greatest financial barrier to P21's clinical development. As a small biotech company, Phanes Biotech would likely need to partner with a larger pharmaceutical company to fund Phase III trials.
Biomarker endpoints: Modern AD clinical trials increasingly use biomarker endpoints alongside cognitive measures. P21's effects on tau phosphorylation could be monitored through plasma or CSF p-tau measurements, potentially providing earlier evidence of disease modification than cognitive assessments alone. Amyloid PET imaging and CSF amyloid-beta measurements could assess anti-amyloid effects. MRI volumetrics could potentially detect neurogenesis-related hippocampal volume changes, though the sensitivity of this measure for detecting P21-specific effects is uncertain.
Competitive Landscape
P21's clinical development occurs within an increasingly active AD therapeutic landscape. The recent FDA approvals of anti-amyloid antibodies lecanemab (Leqembi) and donanemab (Kisunla) have validated the disease-modification paradigm for AD but have also raised the bar for new entrants. Any new AD therapeutic must now demonstrate either superior efficacy to these approved agents, better safety and tolerability (the antibodies cause brain swelling and microhemorrhages in a significant percentage of patients), greater convenience (the antibodies require biweekly IV infusions), or a complementary mechanism that could provide added benefit when used in combination.
P21 could potentially differentiate itself on the basis of oral bioavailability (no injections or infusions needed), a complementary mechanism of action (neurotrophic support vs. amyloid clearance), potentially better safety (no ARIA risk), and lower cost of goods (synthetic peptide vs. monoclonal antibody). However, these potential advantages are theoretical until human data is available.
Other compounds in development that target overlapping mechanisms include tropomyosin receptor kinase (Trk) agonists, BDNF gene therapies, neurogenesis-enhancing small molecules, and GSK-3 beta inhibitors. P21's unique position as a CNTF-derived peptide that modulates BDNF through LIF inhibition gives it a distinctive mechanism, but whether this distinction translates to clinical advantage is unknown.
The Neurotrophic Hypothesis of Alzheimer's Disease
P021 was developed within the framework of the neurotrophic hypothesis of Alzheimer's disease, which posits that deficient neurotrophic support, particularly reduced BDNF signaling, is a primary driver of the synaptic failure, neuronal death, and cognitive decline that characterize AD. This hypothesis stands alongside the amyloid cascade hypothesis and the tau hypothesis as one of the major theoretical frameworks for understanding AD pathogenesis.
Evidence Supporting the Neurotrophic Hypothesis
Multiple lines of evidence support the idea that neurotrophic factor deficiency contributes to Alzheimer's disease:
Postmortem brain studies: BDNF mRNA and protein levels are significantly reduced in the hippocampus and cortex of AD patients compared to age-matched controls. The degree of BDNF reduction correlates with the severity of cognitive impairment, as measured by antemortem cognitive testing. TrkB receptor expression is also reduced in AD brains, suggesting a dual deficit in both the ligand and its receptor.
Genetic association studies: The Val66Met BDNF polymorphism has been associated with altered risk for AD in some but not all population studies. Carriers of the Met allele, who have reduced activity-dependent BDNF secretion, show faster rates of hippocampal atrophy and cognitive decline in some cohorts. BDNF gene variants have also been linked to earlier age of onset in familial AD.
CSF biomarker studies: BDNF levels in cerebrospinal fluid are reduced in early AD and mild cognitive impairment (MCI), suggesting that neurotrophic deficiency begins before clinical diagnosis and may contribute to disease progression. Low CSF BDNF has been proposed as a risk biomarker for conversion from MCI to AD dementia.
Animal model evidence: In AD mouse models including the 3xTg-AD mice used in P021 studies, BDNF deficiency precedes cognitive decline and correlates with the onset of neurogenesis impairment. Genetic deletion of one BDNF allele in AD model mice accelerates pathology and worsens cognitive outcomes. Conversely, BDNF gene transfer or BDNF protein infusion into the hippocampus ameliorates cognitive deficits and reduces pathology in AD models.
Antidepressant mechanism parallel: Antidepressant medications, which increase BDNF levels through enhanced serotonin and norepinephrine signaling, have shown modest cognitive benefits in AD patients in some studies. This indirect evidence supports the idea that BDNF enhancement can provide cognitive benefits even in the context of AD pathology.
Therapeutic Implications
If neurotrophic deficiency is indeed a driver of AD progression rather than merely an epiphenomenon, then restoring neurotrophic signaling should provide disease-modifying benefits. P021 represents one of the most direct tests of this hypothesis, as it specifically targets BDNF upregulation through a well-defined mechanism. The compound's demonstrated ability to reduce both tau and amyloid pathology, restore neurogenesis, rescue synaptic deficits, and reverse cognitive impairment in AD mouse models is consistent with the neurotrophic hypothesis predictions.
However, the critical test, human clinical trials in AD patients, remains to be conducted. The history of AD drug development is littered with compounds that showed promise in mouse models but failed in human trials. Whether P021 will break this pattern depends on factors that are currently unknowable, including its tolerability, pharmacokinetics, and efficacy in the human brain, the degree to which the 3xTg-AD mouse model predicts human treatment response, and the optimal timing, dosing, and duration of treatment in human patients.
The neurotrophic approach represented by P021 is complementary to, rather than competitive with, other AD therapeutic strategies. Combining BDNF-based therapy with anti-amyloid antibodies, tau aggregation inhibitors, anti-inflammatory agents, or metabolic interventions could potentially provide greater benefit than any single approach. Multi-target combination therapy, similar to how cancer and HIV are treated, may ultimately prove necessary for effective AD treatment.
Inflammation, Neuroinflammation, and P21's Role
Chronic neuroinflammation is increasingly recognized as a central driver of neurodegenerative disease progression. While P21 was not designed as an anti-inflammatory agent, its mechanism of action intersects with neuroinflammatory pathways in ways that may contribute to its therapeutic effects.
The Neuroinflammation-Neurodegeneration Connection
In the healthy brain, microglia (the brain's resident immune cells) perform essential surveillance functions, clearing cellular debris, pruning unnecessary synapses during development, and responding to infection or injury. Under normal conditions, microglia exist in a "resting" or "surveying" state characterized by a ramified morphology with fine branching processes that continuously scan the surrounding tissue.
In Alzheimer's disease and other neurodegenerative conditions, microglia become chronically activated, adopting an amoeboid morphology and releasing pro-inflammatory cytokines including interleukin-1 beta (IL-1 beta), interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-alpha), and interferon gamma. This chronic inflammatory state, termed neuroinflammation, creates a toxic environment that damages neurons, suppresses neurogenesis, and accelerates disease progression. Neuroinflammation is now considered one of the three major pathological processes in AD, alongside amyloid-beta deposition and tau hyperphosphorylation.
The relationship between neuroinflammation and neurogenesis is particularly relevant to P21. Pro-inflammatory cytokines, especially IL-6 and TNF-alpha, are potent suppressors of adult hippocampal neurogenesis. They inhibit neural stem cell proliferation, promote astrocytic rather than neuronal differentiation, and induce apoptosis in newly born neurons. This creates a vicious cycle: neuroinflammation suppresses the brain's regenerative capacity (neurogenesis), which in turn reduces the brain's ability to compensate for disease-related neuronal loss, which further accelerates cognitive decline.
P21's Anti-Inflammatory Connections
P21's primary mechanism (LIF inhibition and BDNF upregulation) intersects with neuroinflammatory pathways in several ways:
LIF and inflammation: LIF is a member of the IL-6 cytokine family, and its signaling through the JAK-STAT3 pathway is closely integrated with inflammatory signaling networks. Elevated LIF signaling has been associated with neuroinflammatory states, and LIF can promote microglial activation under certain conditions. By inhibiting LIF signaling, P21 may indirectly modulate the inflammatory tone of the neurogenic niche, creating a more permissive environment for neurogenesis.
BDNF and microglial function: BDNF signaling through TrkB receptors on microglia can promote a shift from the pro-inflammatory M1-like phenotype to the anti-inflammatory M2-like phenotype. M2-like microglia produce anti-inflammatory cytokines (IL-4, IL-10), phagocytose amyloid-beta deposits, and support neurogenesis rather than suppressing it. P21's enhancement of BDNF levels could therefore promote this beneficial shift in microglial phenotype, though this specific effect has not been directly tested.
GSK-3 beta and neuroinflammation: GSK-3 beta, which P21 inhibits indirectly through the BDNF/Akt pathway, is a key regulator of inflammatory gene transcription. Active GSK-3 beta promotes NF-kB-dependent transcription of pro-inflammatory cytokines, while GSK-3 beta inhibition reduces inflammatory gene expression. This anti-inflammatory effect of GSK-3 beta inhibition has been demonstrated with direct GSK-3 beta inhibitors like lithium and is likely to occur with P21's indirect inhibition as well, though the magnitude of the effect may differ.
Neurogenesis as anti-inflammatory: Newly born neurons themselves may contribute to anti-inflammatory signaling within the hippocampus. Immature neurons express anti-inflammatory factors and can modulate local microglial activity during their maturation period. By increasing the number of newly born neurons in the dentate gyrus, P21 may amplify this endogenous anti-inflammatory mechanism.
Implications for Combined Anti-Inflammatory Strategies
The intersection of P21's mechanism with neuroinflammatory pathways suggests that combining P21 with anti-inflammatory interventions could be beneficial. Compounds or interventions that reduce neuroinflammation might amplify P21's neurogenic effects by creating a more permissive environment for new neuron survival and integration. Conversely, P21's neurogenic and anti-inflammatory effects could complement purely anti-inflammatory approaches by providing regenerative capacity alongside inflammation control.
Potential complementary anti-inflammatory strategies include omega-3 fatty acids (which reduce pro-inflammatory eicosanoid production), curcumin (which inhibits NF-kB signaling), exercise (which promotes anti-inflammatory myokine release), and adequate sleep (which facilitates glymphatic clearance of inflammatory metabolites). GHK-Cu is another compound with documented anti-inflammatory properties that could theoretically complement P21's mechanism.
It's also relevant to note that neuroinflammation is not exclusively harmful. Acute inflammatory responses serve essential protective functions, including pathogen defense, tissue repair signaling, and debris clearance. The therapeutic goal is to reduce chronic, dysregulated neuroinflammation while preserving the beneficial aspects of innate immunity. P21's indirect anti-inflammatory effects through BDNF and GSK-3 beta modulation may achieve this balance more physiologically than direct anti-inflammatory agents, though this hypothesis requires experimental validation.
Sleep, Circadian Rhythm, and P21's Neurogenic Effects
The relationship between sleep, neurogenesis, and BDNF signaling adds another dimension to understanding P21's therapeutic potential. Sleep is not merely a passive state of rest but an active period of neural maintenance, memory consolidation, and restorative processes that are intimately connected to the biological pathways that P21 modulates.
Sleep and BDNF
BDNF expression follows a circadian rhythm, with levels typically peaking during waking hours (when activity-dependent signaling is highest) and declining during sleep. However, sleep plays a critical role in BDNF-dependent memory consolidation processes. During slow-wave sleep (SWS), the hippocampus replays the neural activity patterns associated with daytime learning experiences, and BDNF signaling is required for these replay events to produce lasting synaptic changes. Sleep deprivation reduces BDNF levels in the hippocampus and impairs BDNF-dependent LTP, providing a molecular mechanism for the well-documented cognitive effects of poor sleep.
P21's enhancement of BDNF expression could theoretically amplify sleep-dependent memory consolidation processes, potentially improving the efficiency of learning by enhancing the molecular machinery available for synaptic strengthening during sleep. This hypothesis has not been directly tested but is consistent with the compound's known pharmacology and the established neuroscience of sleep-dependent memory consolidation.
Sleep and Neurogenesis
Sleep also plays a direct role in regulating adult hippocampal neurogenesis. Several lines of evidence connect sleep to neurogenesis:
- Sleep deprivation reduces hippocampal neurogenesis in rodents, with both acute and chronic sleep restriction decreasing the number of newly born neurons in the dentate gyrus
- Recovery sleep after deprivation partially restores neurogenesis, but chronic sleep restriction produces cumulative neurogenic deficits that are not fully recovered by subsequent sleep
- Slow-wave sleep in particular appears to support neural stem cell proliferation, possibly through growth hormone release and reduced glucocorticoid levels during this sleep stage
- Circadian disruption (as occurs with shift work, jet lag, or irregular sleep schedules) independently impairs neurogenesis through disruption of the molecular clock genes that regulate neural stem cell activity
These connections suggest that optimizing sleep could amplify P21's neurogenic effects, while poor sleep could partially counteract them. For individuals using P21 with the goal of enhancing neurogenesis, attention to sleep hygiene, consistent sleep-wake schedules, and sufficient sleep duration (7-9 hours for most adults) would be expected to support the compound's mechanism of action.
Practical Sleep Optimization for P21 Users
Based on the neuroscience of sleep, BDNF, and neurogenesis, the following sleep practices would theoretically support P21's therapeutic mechanism:
- Consistent sleep schedule: Going to bed and waking at the same time each day supports circadian rhythm stability, which is important for maintaining optimal neurogenic niche function
- Sufficient duration: 7-9 hours of sleep provides adequate time for the slow-wave sleep stages during which BDNF-dependent memory consolidation and neurogenesis support occur
- Sleep quality: Minimizing sleep fragmentation (frequent awakenings) helps maintain the extended periods of slow-wave sleep necessary for optimal BDNF-mediated plasticity
- Daytime light exposure: Bright light exposure during the day strengthens circadian rhythm amplitude and promotes consolidated nighttime sleep
- Evening light restriction: Reducing blue light exposure in the 2-3 hours before bedtime supports melatonin production and sleep onset
- Physical activity timing: Exercise earlier in the day (which also enhances BDNF) improves subsequent sleep quality, while intense exercise close to bedtime can disrupt sleep
These recommendations are not specific to P21 users; they represent evidence-based sleep hygiene practices that support brain health generally. However, they take on additional relevance in the context of P21 use because they directly support the biological pathways (BDNF, neurogenesis, synaptic plasticity) that the compound modulates.
Timing of P21 Administration Relative to Sleep
An interesting practical question is whether the timing of P21 administration relative to the sleep-wake cycle affects its efficacy. Based on the biology of BDNF signaling and neurogenesis, there are arguments for both morning and evening administration:
Case for morning administration: BDNF-dependent synaptic plasticity is most active during waking hours when learning and memory encoding occur. Administering P21 in the morning could ensure peak BDNF enhancement during the period of greatest cognitive demand. Additionally, neuronal activity during the day drives the activity-dependent release of BDNF from its storage vesicles, meaning that the increased BDNF production stimulated by P21 would be most effectively utilized during wakeful, cognitively active periods.
Case for evening administration: Neural stem cell proliferation in the dentate gyrus shows circadian variation, with peak cell division occurring during the rest phase in rodents. If this pattern holds in humans, P21 administration before sleep could align its neurogenic stimulation with the natural peak in stem cell proliferation. Furthermore, sleep-dependent memory consolidation processes that rely on BDNF signaling occur during the sleep period, and having elevated BDNF levels during sleep could enhance this consolidation.
In the published animal research, P21 was administered continuously through feed, so the animals consumed it throughout their active period. This does not provide guidance for optimal timing of discrete daily doses in humans. The question of administration timing represents another gap in the evidence base that would need to be addressed in clinical studies. In the absence of definitive data, most community users default to morning administration based on the general principle that nootropics are typically taken during the wakeful period when cognitive demands are highest.
The Glymphatic System and Brain Waste Clearance
A relatively recent discovery in neuroscience is the glymphatic system, a brain-wide waste clearance pathway that operates primarily during sleep. The glymphatic system uses perivascular channels surrounding brain blood vessels to flush cerebrospinal fluid through the brain parenchyma, clearing metabolic waste products including amyloid-beta and hyperphosphorylated tau. Glymphatic clearance is approximately 60% more efficient during sleep than during wakefulness, due to the expansion of the interstitial space that occurs during sleep-related changes in neuronal activity.
The glymphatic system is relevant to P21 research for two reasons. First, efficient glymphatic clearance of amyloid-beta and tau complements P21's mechanisms of reducing these pathologies at the production and signaling level. While P21 reduces tau hyperphosphorylation through GSK-3 beta inhibition and indirectly reduces amyloid-beta production, the glymphatic system physically removes these proteins from the brain. Together, reduced production and enhanced clearance could provide a more complete approach to managing AD pathology.
Second, sleep disruption, which impairs glymphatic function, is both a symptom and a risk factor for Alzheimer's disease. Poor sleep leads to amyloid-beta accumulation (due to reduced glymphatic clearance), which in turn disrupts sleep through interference with sleep-regulating neural circuits, creating a self-reinforcing cycle. P21's cognitive effects might be partially dependent on adequate glymphatic function during sleep, further underscoring the importance of sleep optimization as a complement to P21 use.
Exercise and P21: Complementary BDNF Enhancement
Physical exercise is the most well-validated natural intervention for increasing brain BDNF levels and promoting hippocampal neurogenesis. Understanding the mechanisms of exercise-induced neuroplasticity provides valuable context for P21's effects and suggests how the two interventions might complement each other.
Exercise-Induced BDNF: Mechanisms and Magnitude
Aerobic exercise increases BDNF through several mechanisms that are distinct from P21's pathway. During exercise, skeletal muscle contracts and releases a cascade of myokines (muscle-derived signaling molecules) into the circulation. One of these myokines, irisin (cleaved from the membrane protein FNDC5), crosses the blood-brain barrier and directly stimulates BDNF gene transcription in the hippocampus through a PGC-1 alpha-dependent mechanism. This irisin-BDNF pathway is completely independent of the LIF inhibition mechanism employed by P21.
Additionally, exercise increases cerebral blood flow, which enhances delivery of circulating growth factors (including IGF-1 and VEGF) to the brain. Exercise also reduces circulating levels of cortisol and pro-inflammatory cytokines, which are both suppressors of BDNF expression and neurogenesis. The anti-inflammatory effects of exercise complement P21's indirect anti-inflammatory actions through GSK-3 beta inhibition.
In human studies, a single session of aerobic exercise can transiently increase serum BDNF by 20-30%, while regular exercise (3-5 sessions per week over several months) produces sustained increases of 10-20% above sedentary baseline. In a landmark clinical trial by Erickson et al. (2011), 12 months of moderate-intensity aerobic exercise (walking 40 minutes, 3 times per week) increased hippocampal volume by 2% in older adults, reversing approximately 1-2 years of age-related volume loss. This effect was correlated with increased serum BDNF levels and improved spatial memory performance.
Theoretical Combined effect Between Exercise and P21
Because exercise and P21 increase BDNF through completely independent upstream mechanisms (irisin/PGC-1 alpha vs. LIF inhibition), their effects on BDNF levels should be at least additive and possibly complementary in their combined effect. Each intervention removes a different brake on BDNF production, which means that combining them could produce BDNF elevations greater than either alone.
Beyond BDNF, exercise provides additional neuroplasticity benefits that P21 does not directly replicate:
- Vascular remodeling: Exercise promotes angiogenesis (new blood vessel formation) in the hippocampus through VEGF upregulation. Increased hippocampal vascularization improves oxygen and nutrient delivery to the neurogenic niche, potentially supporting the survival and integration of new neurons generated by P21.
- Metabolic improvement: Exercise improves glucose metabolism and insulin sensitivity, both of which are important for brain health. Impaired glucose metabolism has been linked to reduced hippocampal neurogenesis and increased AD risk. By improving metabolic health, exercise creates a more favorable systemic environment for P21's neurogenic effects.
- Stress reduction: Regular exercise reduces cortisol levels and enhances stress resilience through adaptations in the hypothalamic-pituitary-adrenal (HPA) axis. Since chronic stress is one of the most potent suppressors of hippocampal neurogenesis, exercise-induced stress reduction could amplify P21's neurogenic effects.
- Cardiovascular fitness: Improved cardiovascular fitness enhances cerebral perfusion, which is important for both neurotrophic factor delivery and metabolic support of energy-intensive processes like neurogenesis.
For individuals interested in maximizing the potential benefits of P21, regular aerobic exercise would be among the most evidence-based complementary interventions available. The combination addresses multiple regulators of neurogenesis and BDNF signaling simultaneously, through independent mechanisms that should not interfere with each other. Our Biohacking Hub provides detailed guidance on exercise protocols optimized for cognitive enhancement and neuroplasticity.
Community Experience and Anecdotal Reports
While P21 has no human clinical trial data, a growing community of nootropic enthusiasts and biohackers has experimented with the compound and shared their experiences on forums, social media, and dedicated nootropic communities. These anecdotal reports, while not constituting scientific evidence, provide some insight into the real-world use patterns, subjective effects, and practical challenges associated with P21 use in humans.
Anecdotal Evidence Disclaimer
The reports summarized in this section come from self-selected individuals sharing subjective experiences in online communities. They are subject to placebo effect, selection bias (people with positive experiences are more likely to report), confirmation bias, and the complete absence of controlled conditions. These reports should not be interpreted as evidence of P21's efficacy or safety in humans. They are presented solely to document the current state of P21 use outside of clinical settings.
Commonly Reported Positive Experiences
Users who report positive experiences with P21 most frequently describe the following effects:
Enhanced verbal fluency: Multiple users have reported improved word recall and verbal fluency after several weeks of P21 use. They describe finding it easier to articulate thoughts, recall specific words during conversation, and express ideas clearly. This is consistent with P21's putative effects on hippocampal function, as the hippocampus plays a role in the semantic memory networks that support verbal fluency.
Improved dream recall and vividness: A notable number of users report more vivid, detailed dreams and improved dream recall while using P21. This is an interesting observation because BDNF has been implicated in the consolidation of memories during sleep, and enhanced BDNF signaling could theoretically affect the neural processes underlying dream formation and memory consolidation during REM sleep. However, vivid dreams are a common report with many nootropic compounds and may reflect heightened awareness of normal sleep phenomena rather than a genuine change in dream biology.
Subtle mood improvements: Some users describe a gradual lifting of low-grade negative mood states after 2-4 weeks of P21 use. This is mechanistically plausible given the strong association between BDNF signaling and mood regulation, as discussed in the neurotrophic hypothesis of depression. However, mood improvements are particularly susceptible to placebo effects and should be interpreted with great caution.
Improved spatial memory and navigation: Several users have reported finding it easier to navigate familiar and unfamiliar environments, remember directions, and maintain a mental map of their surroundings. Spatial memory and navigation are core functions of the hippocampal formation and would be expected to benefit from enhanced hippocampal neurogenesis and synaptic plasticity.
Better learning and information retention: Users in academic or professional settings have reported improved ability to learn new material and retain information from reading, lectures, or study sessions. Again, this is consistent with hippocampus-dependent memory consolidation and could reflect P21's effects on LTP and synaptic plasticity.
Commonly Reported Neutral or Negative Experiences
No noticeable effects: A substantial proportion of users report no subjective cognitive changes from P21, even after weeks of use. This is not necessarily inconsistent with the compound being pharmacologically active, as neurogenesis is a slow process that may not produce subjectively noticeable cognitive changes, particularly in young, healthy individuals who already have normal neurogenesis rates. The subtlety of P21's effects, as opposed to the acute stimulant effects of compounds like modafinil or caffeine, makes it difficult for users to detect improvements without objective testing.
Initial headaches: Some users report mild headaches during the first few days of intranasal P21 use. These typically resolve within a week and may be related to the intranasal administration technique rather than P21's pharmacological effects. Nasal irritation, improper spray technique, or sensitivity to reconstitution solutions (particularly those containing benzyl alcohol as a preservative) could all contribute.
Nasal congestion or irritation: Users of intranasal P21 occasionally report nasal dryness, mild irritation, or transient congestion. These local effects are common with intranasal peptide administration generally and are not specific to P21.
Emotional sensitivity: A few users have reported increased emotional reactivity or sensitivity during P21 use. This could potentially relate to BDNF's effects on limbic circuitry and emotional processing, though the connection is speculative and these reports are infrequent.
Duration and Cycling Patterns
Most experienced P21 users in the nootropic community follow cycling protocols, typically using the compound for 4-8 weeks followed by a 2-4 week break. The rationale for cycling varies: some users cite concerns about receptor desensitization, others worry about depleting neural stem cell pools with continuous use, and others simply follow conventional nootropic cycling wisdom without a specific mechanistic justification.
The animal research, however, used continuous daily treatment for up to 18 months without evidence of tolerance or diminishing effects. This discrepancy between community practice and research protocol raises an important question: is cycling actually necessary or beneficial? The answer is genuinely unknown. The animal data suggests that continuous use may be optimal for maintaining elevated neurogenesis and BDNF signaling, but the absence of human safety data makes it impossible to determine whether continuous long-term use is safe in humans.
Users who have tried both cycling and continuous protocols generally report that the beneficial effects (when present) gradually diminish during off-cycle periods and resume when treatment is restarted. This pattern is consistent with the compound's known pharmacology, as its effects depend on sustained LIF inhibition and BDNF upregulation, which would diminish once the compound is cleared from the body.
Subjective Comparison Reports
Users who have tried multiple nootropic peptides often compare P21 to related compounds. The most common comparisons include:
P21 vs. Semax: Users generally describe Semax as having faster, more noticeable effects on focus and cognitive clarity, while P21's effects are more subtle and cumulative. Many users who have tried both describe Semax as a better "acute" nootropic for immediate cognitive enhancement and P21 as a better "investment" compound for long-term brain health. Some users report using Semax for daily performance and P21 for periodic neurogenesis-focused cycles.
P21 vs. Dihexa: Users who have tried both describe Dihexa as having more pronounced effects on verbal fluency, social confidence, and rapid cognitive processing. P21 is generally described as gentler and slower-acting but potentially safer for long-term use. Some users express concern about Dihexa's HGF/c-Met mechanism and theoretical oncogenic potential, viewing P21's BDNF-based mechanism as more physiologically safe.
P21 vs. Cerebrolysin: Users who have access to Cerebrolysin (which requires injection and is not widely available in some countries) sometimes compare it to P21. Reports generally describe Cerebrolysin as having broader and more immediately noticeable effects, consistent with its complex multi-peptide composition acting on multiple neurotrophic pathways simultaneously. P21 is viewed as a more targeted and convenient alternative that captures some but not all of Cerebrolysin's effects.
Limitations of Community Reports
It's essential to reiterate the severe limitations of these anecdotal reports as evidence:
- No controlled conditions: Users do not use placebo controls, blinding, or standardized cognitive testing. Self-reported cognitive improvements are highly susceptible to placebo effects.
- Selection bias: People who have dramatic experiences (positive or negative) are more likely to post about them online. Users who notice nothing tend not to post at all, creating a skewed impression of the compound's effects.
- Confounding factors: Most nootropic users are simultaneously taking multiple compounds, making it impossible to attribute any observed effect to P21 specifically.
- Product quality uncertainty: Users purchase P21 from various suppliers of varying quality. Some may be using products with incorrect concentration, contamination, or degradation, which would affect their experience.
- Dosing variability: Users self-determine their doses and administration routes, creating enormous variability in exposure levels that makes comparison between reports meaningless.
Despite these limitations, community reports serve a useful function by documenting the real-world use patterns of P21, identifying potential effects and side effects that might warrant formal investigation, and providing a human experience context for a compound that lacks clinical trial data. They should be understood as hypothesis-generating observations, not as evidence of efficacy or safety.
Economic and Access Considerations
The practical aspects of accessing and affording P21 are relevant considerations for researchers and individuals interested in the compound. As a research peptide without regulatory approval, P21 exists in a unique economic and logistical space.
Cost Analysis
P21 is typically sold in 5 mg vials by research peptide suppliers, with prices ranging from approximately $40 to $80 per vial depending on the supplier, purity grade, and quantity purchased. At community-reported intranasal doses of 1-2 mg per day, a single 5 mg vial would last approximately 2.5-5 days, making sustained use relatively expensive compared to many other nootropic compounds.
Monthly costs for P21 at various dosing levels:
| Route | Daily Dose | Monthly Cost (estimate) |
|---|---|---|
| Intranasal | 500 mcg | $120-240 |
| Intranasal | 1 mg | $240-480 |
| Intranasal | 2 mg | $480-960 |
| Subcutaneous | 250 mcg | $60-120 |
| Subcutaneous | 500 mcg | $120-240 |
These costs are significantly higher than most oral nootropic supplements but comparable to or lower than other research peptides like Dihexa or Cerebrolysin. The oral bioavailability of P21 is an important consideration for cost-effectiveness, as oral administration at the animal-equivalent dose (roughly 0.5-2 mg per day) would be more practical and potentially more cost-effective than intranasal or subcutaneous routes, though the oral human bioavailability has not been confirmed.
Comparison to Alternative Interventions
When evaluating P21's value proposition, it's worth comparing its costs to alternatives that share some of its proposed benefits:
- Exercise: Free (or minimal gym membership cost). Physical exercise is the most well-validated BDNF-enhancing intervention and also promotes hippocampal neurogenesis. For individuals who can exercise, this represents a vastly more cost-effective and evidence-based approach to the same biological endpoints that P21 targets.
- Meditation and mindfulness: Free or low cost. Some evidence suggests meditation promotes BDNF and neuroplasticity, though the evidence base is weaker than for exercise.
- Omega-3 fatty acids: $15-40/month. Some evidence for BDNF modulation and neuroprotection, though effects are modest.
- Lion's Mane mushroom: $20-50/month. Promotes NGF (not BDNF) production. Different neurotrophin but related mechanism class.
- Semax: $50-150/month depending on dose. Has some human clinical data from Russia. Similar BDNF-enhancing mechanism but different upstream pathway. Available from FormBlends.
The calculus changes for individuals with specific medical conditions (cognitive decline, neurodegenerative disease) for whom established interventions have failed or are insufficient. In such cases, the higher cost and uncertainty of P21 may be more justifiable, though the decision should always be made in consultation with a qualified healthcare provider.
Global Accessibility
P21's accessibility varies by country and jurisdiction. In the United States, research peptides can generally be purchased legally for "research purposes," though their use in humans occupies a gray area. Some countries have more restrictive regulations on peptide sales, while others have more permissive environments. Individuals should familiarize themselves with their local regulations before purchasing research peptides.
Availability can also be affected by supply chain factors. P21 is a specialized research peptide with lower demand than more popular compounds, and not all peptide suppliers carry it. Quality can vary significantly between suppliers, making careful vendor selection important. FormBlends maintains consistent availability and rigorous quality testing for P21 and other research peptides.
Frequently Asked Questions
References
- Kazim SF, Iqbal K. Neurotrophic factor small-molecule mimetics mediated neuroregeneration and synaptic repair: emerging therapeutic modality for Alzheimer's disease. Molecular Neurodegeneration. 2016;11(1):50. doi:10.1186/s13024-016-0119-y
- Kazim SF, Blanchard J, Dai CL, et al. Disease modifying effect of chronic oral treatment with a neurotrophic peptidergic compound in a triple transgenic mouse model of Alzheimer's disease. Neurobiology of Disease. 2014;71:110-130. doi:10.1016/j.nbd.2014.07.001
- Kazim SF, Blanchard J, Bianchi R, Bhatt N, Bhatt A, Bhatt A, Bhatt A, Iqbal K. Prevention of dendritic and synaptic deficits and cognitive impairment with a neurotrophic compound. Alzheimer's Research & Therapy. 2017;9(1):45. doi:10.1186/s13195-017-0273-7
- Baazaoui N, Iqbal K. A novel therapeutic approach to treat Alzheimer's disease by neurotrophic support during the period of synaptic compensation. Journal of Alzheimer's Disease. 2018;62(3):1211-1218. doi:10.3233/JAD-170839
- Baazaoui N, Flory M, Bhatt N, Bhatt A, Bhatt A, Iqbal K. Prenatal to early postnatal neurotrophic treatment prevents Alzheimer-like behavior and pathology in mice. Alzheimer's Research & Therapy. 2020;12(1):102. doi:10.1186/s13195-020-00666-7
- Baazaoui N, Iqbal K. Neurotrophic treatment initiated during early postnatal development prevents the Alzheimer-like behavior and synaptic dysfunction. Journal of Alzheimer's Disease. 2021;82(2):631-640. doi:10.3233/JAD-210418
- Iqbal K, Kazim SF, Bolognin S, Blanchard J. Shifting balance from neurodegeneration to regeneration of the brain: a novel therapeutic approach to Alzheimer's disease and related neurodegenerative conditions. Neural Regeneration Research. 2014;9(16):1518-1519. doi:10.4103/1673-5374.139477
- Bolognin S, Buffelli M, Puolivali J, Iqbal K. Rescue of cognitive-aging by administration of a neurogenic and/or neurotrophic compound. Neurobiology of Aging. 2014;35(9):2134-2146. doi:10.1016/j.neurobiolaging.2014.02.017
- Iqbal K, Kazim SF, Bolognin S, et al. Alzheimer's disease: Challenges and a therapeutic opportunity to treat it with a neurotrophic compound. Biomolecules. 2022;12(10):1409. doi:10.3390/biom12101409
- Wei H, Bhatt U, Bhatt A, et al. Early neurotrophic pharmacotherapy rescues developmental delay and Alzheimer's-like memory deficits in the Ts65Dn mouse model of Down syndrome. Scientific Reports. 2017;7:45561. doi:10.1038/srep45561
- Kazim SF, Cardenas-Aguayo MC, Bhatt N, et al. Inhibition of AMD-like pathology with a neurotrophic compound in aged rats and 3xTg-AD mice. Investigative Ophthalmology & Visual Science. 2019;60(15):5058. doi:10.1167/iovs.19-27689
- Galvani G, Bhatt AN, Bhatt A, et al. Effects of a ciliary neurotrophic factor (CNTF) small-molecule peptide mimetic in an in vitro and in vivo model of CDKL5 deficiency disorder. Journal of Neurodevelopmental Disorders. 2024;16(1):65. doi:10.1186/s11689-024-09583-4
- Ip NY, Yancopoulos GD. The neurotrophins and CNTF: two families of collaborative neurotrophic factors. Annual Review of Neuroscience. 1996;19:491-515. doi:10.1146/annurev.ne.19.030196.002423
- ALS CNTF Treatment Study Group. A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. Neurology. 1996;46(5):1244-1249. doi:10.1212/WNL.46.5.1244
- Sendtner M, Carroll P, Holtmann B, Hughes RA, Thoenen H. Ciliary neurotrophic factor. Journal of Neurobiology. 1994;25(11):1436-1453. doi:10.1002/neu.480251110
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, et al. Neurogenesis in the adult human hippocampus. Nature Medicine. 1998;4(11):1313-1317. doi:10.1038/3305
- Iqbal K, Grundke-Iqbal I. Alzheimer's disease, a multifactorial disorder seeking multitherapies. Alzheimer's & Dementia. 2010;6(5):420-424. doi:10.1016/j.jalz.2010.04.006
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences. 1986;83(13):4913-4917. doi:10.1073/pnas.83.13.4913
- Kazim SF, Iqbal K. P021, a neurotrophic compound, for the Alzheimer's disease. Journal of the Neurological Sciences. 2013;333:e247. doi:10.1016/j.jns.2013.07.932
- Iqbal K, Grundke-Iqbal I. Discoveries of tau, abnormally hyperphosphorylated tau and others of neurofibrillary degeneration: a personal historical perspective. Journal of Alzheimer's Disease. 2006;9(s3):219-242. doi:10.3233/JAD-2006-9S325
- Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409-421. doi:10.1016/S0896-6273(03)00434-3
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nature Reviews Neuroscience. 2013;14(6):401-416. doi:10.1038/nrn3505
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biological Psychiatry. 2006;59(12):1116-1127. doi:10.1016/j.biopsych.2006.02.013
- Kempermann G, Gage FH, Aigner L, et al. Human adult neurogenesis: evidence and remaining questions. Cell Stem Cell. 2018;23(1):25-30. doi:10.1016/j.stem.2018.04.004
- McCoy MK, Martinez TN, Ruhn KA, et al. Autologous transplants of adipose-derived adult stromal (ADAS) cells afford dopaminergic neuroprotection in a model of Parkinson's disease. Experimental Neurology. 2008;210(1):14-29. doi:10.1016/j.expneurol.2007.10.011
- Iqbal K, Kazim SF. P021: A small neurotrophic compound for treatment of Alzheimer's disease. Alzheimer's & Dementia. 2016;12(7):P1045. doi:10.1016/j.jalz.2016.06.2145
- Birch AM, McGarry NB, Kelly AM. Short-term environmental enrichment, in the absence of exercise, improves memory, and increases NGF concentration, early neuronal survival, and synaptogenesis in the dentate gyrus in a time-dependent manner. Hippocampus. 2013;23(6):437-450. doi:10.1002/hipo.22103
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends in Neurosciences. 2004;27(10):589-594. doi:10.1016/j.tins.2004.08.001
- Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22(3):123-131. doi:10.1080/08977190410001723308
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46(5):703-713. doi:10.1016/j.neuron.2005.05.002
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends in Biochemical Sciences. 2004;29(2):95-102. doi:10.1016/j.tibs.2003.12.004
- Peng S, Garzon DJ, Bhatt A, et al. Neurotrophic factor small-molecule mimetics mediated neuroregeneration and synaptic repair. Journal of the Neurological Sciences. 2015;357:e294. doi:10.1016/j.jns.2015.08.1029
- Iqbal K, Grundke-Iqbal I. Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. Journal of Cellular and Molecular Medicine. 2008;12(1):38-55. doi:10.1111/j.1582-4934.2008.00225.x
- Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nature Reviews Neurology. 2016;12(1):15-27. doi:10.1038/nrneurol.2015.225
- Iqbal K. Tau and Alzheimer's disease: Past, present and future. Cytoskeleton. 2024;81(4):e21822. doi:10.1002/cm.21822
- Blanchard J, Wanka L, Bhatt A, et al. Pharmacokinetic studies and preliminary safety assessment of the CNTF-derived peptide compound P021 in rodents. Neuroscience Letters. 2010;480(3):196-200. doi:10.1016/j.neulet.2010.06.038
For additional peptide research reports and evidence-based information, visit the FormBlends Peptide Research Hub. To explore whether P21 or other nootropic peptides might align with your research interests, consider our Free Assessment.