Executive Summary
Choosing the right compounding pharmacy for peptide therapy is one of the most consequential decisions a patient or prescriber will ever make. The difference between a high-quality compounder and a substandard one isn't just academic - it can mean the difference between an effective, safe treatment and a contaminated product that lands someone in the hospital.
Key Takeaways
- 503B outsourcing facilities - offer stronger regulatory oversight than 503A pharmacies, with FDA registration, cGMP compliance, and mandatory adverse event reporting
- PCAB accreditation - adds an additional layer of quality verification through independent on-site surveys
- Every injectable peptide - should come with documentation of potency testing, sterility testing, and endotoxin testing at minimum
- Third-party COAs - from independent labs provide the strongest quality assurance
The compounding pharmacy industry exists in a unique regulatory space. Unlike conventional pharmacies that simply dispense FDA-approved medications, compounding pharmacies create customized preparations - mixing, combining, or altering drug ingredients to produce formulations tailored to individual patient needs. For peptides specifically, this means reconstituting lyophilized powders, preparing sterile injectable solutions, and sometimes combining multiple peptide compounds into single formulations.
This guide was written because the world of peptide compounding has grown increasingly complex. The explosion of interest in GLP-1 receptor agonists like semaglutide and tirzepatide has drawn hundreds of new compounding operations into the market, and not all of them meet the quality standards that injectable medications demand. The FDA issued more than 50 warning letters to GLP-1 compounders in September 2025 alone, signaling serious concerns about quality, safety, and misleading marketing practices across the industry.
Understanding the regulatory framework is your first line of defense. Federal law creates two distinct categories of compounding pharmacies - Section 503A and Section 503B facilities - each with dramatically different oversight requirements, testing standards, and operational capabilities. A 503A pharmacy operates under state board supervision and compounds medications based on individual patient prescriptions. A 503B outsourcing facility registers with the FDA, follows current Good Manufacturing Practices (cGMP), and can produce larger batches without patient-specific prescriptions.
Beyond the regulatory classification, quality markers like PCAB accreditation, USP 797 compliance, third-party testing programs, and transparent Certificates of Analysis (COAs) separate the best compounding pharmacies from the rest. This report will walk you through every one of these quality indicators, teach you how to read a COA, explain what questions to ask your pharmacy, and identify the red flags that should send you looking elsewhere.
The stakes are real. In 2012, the New England Compounding Center (NECC) tragedy killed 64 people and sickened nearly 800 when contaminated steroid injections caused a nationwide fungal meningitis outbreak. That disaster reshaped compounding regulation in the United States and led directly to the creation of the 503B outsourcing facility category under the Drug Quality and Security Act of 2013. More than a decade later, the lessons from NECC remain painfully relevant as the peptide compounding market expands at unprecedented speed.
Whether you're a patient evaluating a compounding pharmacy for your first peptide prescription, a clinician selecting a compounding partner for your practice, or a researcher trying to understand the quality differences between suppliers, this guide provides the evidence-based framework you need to make an informed decision. We've reviewed FDA enforcement data, USP compounding standards, PCAB accreditation requirements, published contamination studies, and real-world warning letters to give you a complete picture of what separates a trustworthy compounding pharmacy from a risky one.
KEY TAKEAWAYS
503B outsourcing facilities offer stronger regulatory oversight than 503A pharmacies, with FDA registration, cGMP compliance, and mandatory adverse event reporting. PCAB accreditation adds an additional layer of quality verification through independent on-site surveys. Every injectable peptide should come with documentation of potency testing, sterility testing, and endotoxin testing at minimum. Third-party COAs from independent labs provide the strongest quality assurance. Red flags include pharmacies that can't provide testing documentation, make claims about being "FDA-approved," sell without prescriptions, or offer prices dramatically below market rates.
What Is Compounding? History, Purpose, and Legal Framework
The Origins of Pharmaceutical Compounding
Pharmacy compounding is as old as pharmacy itself. Before the rise of mass-manufactured pharmaceuticals in the mid-twentieth century, virtually all medications were compounded. Pharmacists - or apothecaries, as they were known historically - would receive a physician's prescription and prepare the medication from raw ingredients right there in the pharmacy. Every pill, every tincture, every ointment was a compounded preparation.
The pharmaceutical manufacturing revolution changed that. By the 1950s and 1960s, large-scale drug manufacturers could produce standardized dosage forms - tablets, capsules, pre-filled syringes - with a consistency and efficiency that individual pharmacies couldn't match. The FDA's regulatory framework evolved to oversee these manufacturers, requiring extensive clinical trials, Good Manufacturing Practices, and post-market surveillance. Compounding didn't disappear, but it shrank to a niche practice serving patients whose needs couldn't be met by commercially available products.
Today, compounding serves several legitimate and essential purposes. Patients who need a medication in a different dosage form - a liquid instead of a tablet for someone who can't swallow pills, for instance - rely on compounding pharmacies. Patients with allergies to inactive ingredients like dyes, preservatives, or fillers in commercial products need compounded alternatives. Pediatric patients often need medications in smaller doses or more palatable formulations than manufacturers produce. And when commercial drug shortages occur, compounding pharmacies fill critical gaps in the drug supply.
For peptide therapy specifically, compounding plays a particularly significant role. Many therapeutic peptides are not available as FDA-approved commercial products, or they may be available only at specific doses that don't match every patient's clinical needs. Peptides like BPC-157, CJC-1295/Ipamorelin, and Thymosin Alpha-1 are available primarily through compounding pharmacies. Even FDA-approved peptides like semaglutide may be compounded during periods of drug shortage, subject to specific regulatory conditions.
The Legal Framework: Federal Food, Drug, and Cosmetic Act
The legal authority for pharmacy compounding in the United States derives from the Federal Food, Drug, and Cosmetic Act (FD&C Act), as amended by several subsequent pieces of legislation. The most significant of these amendments came in response to tragedies that exposed gaps in the regulatory framework.
The Food and Drug Administration Modernization Act of 1997 (FDAMA) added Section 503A to the FD&C Act, establishing the first federal framework for pharmacy compounding. Section 503A provided exemptions from certain FDA requirements - specifically, the new drug approval process, adequate directions for use labeling, and current Good Manufacturing Practice (cGMP) requirements - for pharmacies engaged in traditional compounding activities.
These exemptions came with conditions. To qualify under Section 503A, a pharmacy had to compound based on receipt of a valid prescription for an identified individual patient. The compounded drug couldn't be a copy of a commercially available product (with limited exceptions). The pharmacy couldn't advertise or promote the compounding of specific drug products. And the compounding had to be performed by a licensed pharmacist in a state-licensed pharmacy.
For over 15 years, Section 503A was the primary federal framework governing compounding. But the system had a fatal weakness: enforcement was largely left to state boards of pharmacy, and the standards varied dramatically from state to state. Some states had rigorous inspection programs and strict quality requirements. Others had minimal oversight. And a growing number of compounding operations were functioning more like drug manufacturers than traditional pharmacies - producing large quantities of medications without patient-specific prescriptions and distributing them across state lines.
The NECC Disaster and the Drug Quality and Security Act
In September 2012, the U.S. Centers for Disease Control and Prevention began investigating an outbreak of fungal meningitis among patients who had received epidural steroid injections. The investigation traced the contamination to three lots of preservative-free methylprednisolone acetate produced by the New England Compounding Center (NECC), a compounding pharmacy in Framingham, Massachusetts.
The scale of the outbreak was staggering. NECC had distributed contaminated doses to 75 medical facilities across 23 states. Approximately 14,000 patients received injections from the contaminated lots between May and September 2012. By the time the outbreak was contained, 798 people had been sickened and 64 had died, making it the largest public health crisis ever caused by a contaminated pharmaceutical product in the United States (CDC, 2015).
The subsequent investigation revealed an operation riddled with safety violations. NECC's owner, Barry Cadden, had authorized shipments of medications before sterility testing was confirmed. When tests came back showing contamination, the company failed to notify customers. NECC compounded drugs with expired ingredients and routinely dispensed medications in bulk without valid prescriptions, even using fictional names and celebrity names like "Michael Jackson" and "Diana Ross" on fake prescriptions (DOJ, 2017). Cadden was convicted of racketeering and later pleaded no contest to 11 counts of involuntary manslaughter in Michigan, where 11 victims died.
The NECC tragedy made clear that the existing regulatory framework was inadequate. Congress responded by passing the Drug Quality and Security Act (DQSA) in November 2013, which added Section 503B to the FD&C Act. This new section created a voluntary category called "outsourcing facilities" - compounding operations that would register with the FDA, submit to regular FDA inspections, follow cGMP requirements, and report adverse events, in exchange for the ability to compound without patient-specific prescriptions and distribute products in larger quantities.
The DQSA was a watershed moment for compounding regulation. It created a two-tier system that persists today: 503A pharmacies operating under traditional state oversight, and 503B outsourcing facilities operating under federal FDA oversight. Understanding the differences between these two tiers is essential for anyone evaluating a compounding pharmacy for peptide therapy.
HISTORICAL CONTEXT
Before the DQSA, the FDA had limited authority to regulate compounding pharmacies. The agency could take action against compounders only after problems occurred - a reactive approach that left patients vulnerable. The 503B framework shifted the paradigm toward proactive oversight, with registered facilities subject to FDA inspection before problems arise. However, some critics argue that the voluntary nature of 503B registration means that many large-scale compounders continue to operate under the less rigorous 503A framework (Gudeman et al., 2013).
State vs. Federal Regulation: A Layered System
One of the most important things to understand about compounding pharmacy regulation is that it's a layered system. Federal law establishes the baseline framework, but state laws and regulations add additional requirements that vary significantly from jurisdiction to jurisdiction.
Every state has its own board of pharmacy that licenses and oversees compounding pharmacies operating within its borders. Some states have adopted compounding regulations that exceed federal requirements. California, for example, has particularly stringent requirements for sterile compounding, including mandatory facility inspections and detailed quality assurance protocols. Texas requires compounding pharmacies to maintain extensive documentation of their quality control procedures. Florida has established specific licensing categories for sterile compounding pharmacies.
Other states have less developed regulatory frameworks, with minimal inspection programs and limited quality requirements. This variability means that a pharmacy licensed in one state might not meet the standards expected in another state. For patients and prescribers, this underscores the importance of looking beyond simple licensure status when evaluating a compounding pharmacy.
The interplay between state and federal regulation also creates complexity around interstate distribution. Under Section 503A, compounding pharmacies are generally limited in their ability to distribute compounded preparations across state lines, though the specifics vary by state and the FDA has exercised enforcement discretion in this area. Section 503B facilities, by contrast, can distribute their products nationally because they're registered with the FDA and subject to federal oversight.
For peptide compounding specifically, this means that a patient in one state might receive their compounded peptides from a 503B facility located in another state. As long as the facility maintains its FDA registration and complies with cGMP requirements, this is entirely legal and, in many cases, may provide access to higher-quality compounding than what's available locally.
The Role of USP Standards
The United States Pharmacopeia (USP) plays a critical role in setting quality standards for compounding. USP is a nonprofit scientific organization that develops and publishes standards for medicines, food ingredients, and dietary supplements. Its compounding standards are particularly important because they establish the technical requirements that compounding pharmacies must follow.
Three USP chapters are especially relevant to peptide compounding:
- USP <795> - Pharmaceutical Compounding: Nonsterile Preparations. This chapter covers the compounding of medications that don't need to be sterile, such as oral preparations, topical creams, and capsules.
- USP <797> - Pharmaceutical Compounding: Sterile Preparations. This is the chapter most relevant to peptide compounding, as most peptides are administered by injection and therefore require sterile preparation. USP 797 sets detailed requirements for personnel training, facility design, environmental monitoring, and quality testing.
- USP <800> - Hazardous Drugs: Handling in Healthcare Settings. This chapter applies when compounding pharmacies handle drugs classified as hazardous, establishing requirements for protective equipment, engineering controls, and waste disposal.
While USP standards are not federal law in themselves, they're incorporated by reference into federal and state regulations. The FDA expects 503B outsourcing facilities to follow USP standards as part of their cGMP obligations. State boards of pharmacy typically require compliance with applicable USP chapters as a condition of licensure. And accreditation bodies like PCAB explicitly evaluate compliance with USP standards during their accreditation surveys.
We'll examine USP 797 requirements in detail in a later section, but for now, the key point is this: USP standards represent the minimum quality requirements for sterile compounding. A pharmacy that isn't following USP 797 shouldn't be compounding injectable peptides, period.
503A vs 503B Regulatory Framework
The distinction between Section 503A and Section 503B of the Federal Food, Drug, and Cosmetic Act defines two fundamentally different models of compounding pharmacy operation. Understanding these differences is essential for evaluating the quality and safety of any compounded peptide product.
Section 503A: Traditional Compounding Pharmacies
Section 503A pharmacies represent the traditional model of pharmacy compounding. These are typically brick-and-mortar pharmacies - often community pharmacies or specialty compounding pharmacies - that prepare medications based on individual patient prescriptions. They operate primarily under state board of pharmacy oversight and are exempt from three major FDA requirements: the new drug approval process, cGMP requirements, and adequate directions for use labeling.
To qualify for these exemptions, a 503A pharmacy must meet several conditions:
- Patient-specific prescriptions: The pharmacy must compound based on the receipt of a valid prescription for an individually identified patient. This is the foundational requirement - 503A pharmacies cannot produce drugs in anticipation of receiving prescriptions (with limited exceptions for limited quantities based on established prescription histories).
- Licensed pharmacist: Compounding must be performed by or under the direct supervision of a licensed pharmacist in a state-licensed pharmacy or federal facility.
- No copies of commercial products: The pharmacy generally cannot compound a drug that is essentially a copy of a commercially available drug product, unless that commercial product appears on the FDA's drug shortage list.
- No advertising of specific compounds: The pharmacy cannot advertise or promote the compounding of any particular drug, class of drug, or type of drug. This provision has been the subject of constitutional challenges and its enforcement has varied.
- Ingredient sourcing: Active ingredients must comply with USP or National Formulary monograph standards, be manufactured by an FDA-registered establishment, and be accompanied by valid certificates of analysis.
What 503A pharmacies are NOT required to do is equally revealing:
- They are not required to register with the FDA
- They are not required to follow current Good Manufacturing Practices (cGMP)
- They are not required to report adverse events to the FDA
- They are not subject to routine FDA inspections (though the FDA can inspect them for cause)
- They are not required to perform potency, sterility, or endotoxin testing on every batch (though USP 797 may require some testing depending on risk level)
This doesn't mean 503A pharmacies are unregulated. They're subject to state board of pharmacy oversight, which includes licensing requirements, periodic inspections, and complaint-based investigations. Many 503A pharmacies voluntarily exceed minimum requirements, maintaining rigorous quality programs, performing extensive testing, and achieving PCAB accreditation. But the baseline regulatory requirements for 503A pharmacies are significantly less demanding than those for 503B facilities.
Section 503B: Outsourcing Facilities
Section 503B outsourcing facilities were created by the Drug Quality and Security Act of 2013 as a direct response to the NECC tragedy. They represent a middle ground between traditional compounding pharmacies and full-scale drug manufacturers, with regulatory requirements that are more stringent than 503A but less burdensome than the complete New Drug Application (NDA) process.
Key requirements for 503B outsourcing facilities include:
- FDA registration: Every 503B facility must register with the FDA and provide information about the drugs it compounds. This registration puts the facility on the FDA's radar for inspection and oversight.
- Current Good Manufacturing Practices (cGMP): 503B facilities must comply with cGMP requirements, which cover every aspect of the manufacturing process - from facility design and equipment maintenance to personnel training and quality control testing. This is the same framework that governs conventional drug manufacturers.
- FDA inspections: Registered 503B facilities are subject to routine FDA inspections, not just for-cause investigations. The FDA maintains a risk-based inspection schedule, with higher-risk facilities inspected more frequently.
- Adverse event reporting: 503B facilities must report serious adverse events to the FDA within 15 calendar days. This requirement creates a feedback loop that helps identify quality problems before they become widespread.
- Product labeling: Compounded drugs from 503B facilities must include specific labeling information, including a statement that the product is a compounded preparation and the name of the outsourcing facility.
- No patient-specific prescriptions required: Unlike 503A pharmacies, 503B facilities can compound drugs without individual patient prescriptions. This allows them to produce drugs in advance, maintain inventory, and supply healthcare facilities directly.
Head-to-Head Comparison
| Feature | 503A Pharmacy | 503B Outsourcing Facility |
|---|---|---|
| Primary oversight | State board of pharmacy | FDA (federal) |
| FDA registration required | No | Yes |
| cGMP compliance required | No | Yes |
| Routine FDA inspections | No (for-cause only) | Yes (risk-based schedule) |
| Adverse event reporting to FDA | Not required | Required (15-day deadline) |
| Patient-specific prescription needed | Yes | No |
| Can supply healthcare facilities | Limited | Yes |
| Batch production | Limited anticipatory compounding | Large-scale batch production allowed |
| Potency testing per batch | Not federally required | Required under cGMP |
| Sterility testing per batch | Varies by state/USP risk level | Required under cGMP |
| Endotoxin testing per batch | Varies by state/USP risk level | Required under cGMP |
| Stability testing | Generally not required | Required under cGMP |
| Product labeling requirements | State-specific | Federal requirements apply |
| Interstate distribution | Limited/varies by state | Nationwide distribution permitted |
Which Is Better for Peptide Compounding?
The question of whether a 503A or 503B pharmacy is "better" for peptide compounding doesn't have a simple answer, because quality varies enormously within each category. A well-run 503A pharmacy with PCAB accreditation, voluntary third-party testing, and a strong quality assurance program may produce higher-quality compounded peptides than a poorly managed 503B facility that's skating by on minimum compliance.
That said, the structural advantages of the 503B model are real and significant for peptide compounding:
Testing requirements are non-negotiable. Under cGMP, every batch of a compounded drug from a 503B facility must be tested for potency, sterility, and endotoxin content before release. This isn't optional or dependent on the facility's own quality judgment - it's a regulatory requirement subject to FDA verification. For injectable peptides, where contamination can cause serious harm, this mandatory testing provides a critical safety net.
Process validation is required. 503B facilities must validate their compounding processes, demonstrating that their procedures consistently produce products that meet quality specifications. This includes validating sterilization methods, testing environmental controls, and documenting that every step in the process works as intended. Process validation is one of the most labor-intensive and expensive aspects of cGMP compliance, but it's also one of the most valuable for ensuring consistent product quality.
FDA oversight creates accountability. The FDA's inspection program for 503B facilities means that quality claims can be verified independently. When an FDA inspector walks through a 503B facility, they review production records, examine testing results, evaluate facility conditions, and interview personnel. The findings are documented in inspection reports that are publicly available, giving patients and prescribers an independent window into the facility's operations.
Adverse event reporting catches problems early. The mandatory adverse event reporting requirement for 503B facilities creates a systematic way to identify quality problems. If multiple patients experience adverse reactions to a compounded product, the reports flow to the FDA's adverse event database, where patterns can be identified and investigated. Without this reporting requirement, problems at 503A pharmacies may go undetected until a serious outbreak occurs.
For these reasons, many clinicians and patients prefer 503B outsourcing facilities for compounded peptides, particularly for peptides that are administered by injection. The additional regulatory oversight provides a margin of safety that's especially valuable for products that bypass the body's natural defense barriers when injected subcutaneously or intramuscularly.
However, 503A pharmacies retain important advantages in certain situations. They can provide more personalized compounding - adjusting doses, combining medications, and tailoring formulations to individual patient needs in ways that 503B batch production may not accommodate. They often have a closer relationship with the prescribing physician and the patient. And for patients who need unusual combinations or non-standard dosing, a skilled 503A pharmacy may be the only option.
The best approach is to look beyond the regulatory classification and evaluate each pharmacy on its actual quality practices. Regardless of whether a pharmacy operates under 503A or 503B, the quality indicators we'll discuss in the following sections - USP compliance, accreditation, testing programs, and transparency - are the most reliable predictors of product quality.
CLINICAL PERSPECTIVE
When selecting a compounding pharmacy for peptide prescriptions, clinicians should request documentation of the pharmacy's testing protocols, review recent COAs for the specific peptides they plan to prescribe, and verify the pharmacy's regulatory status and any accreditations. For practices that prescribe peptides regularly, establishing a relationship with a 503B outsourcing facility - or a PCAB-accredited 503A pharmacy with a strong sterile compounding program - provides the most reliable quality assurance for patients. The GLP-1 compounding pharmacy guide offers additional guidance specific to semaglutide and tirzepatide compounding.
The Gray Area: 503A Pharmacies Operating at Scale
One of the ongoing tensions in compounding regulation involves 503A pharmacies that operate at a scale more consistent with manufacturing than traditional compounding. Some 503A pharmacies compound thousands of doses per month, distribute products across multiple states, and market their services directly to consumers through sophisticated websites and social media campaigns. These operations may technically qualify for 503A exemptions if they maintain patient-specific prescriptions, but their scale and distribution patterns raise questions about whether state board oversight alone is sufficient.
The FDA has expressed concern about this gray area repeatedly. In 2023, the agency issued draft guidance permitting 503A pharmacies to purchase compounded medications from 503B facilities for dispensing to patients - an acknowledgment that the boundary between the two categories had become blurred. And the 2024 proposed rule on demonstrable difficulties for compounding signaled the FDA's intent to tighten restrictions on what can be compounded under both 503A and 503B frameworks.
For patients and prescribers, the practical implication is straightforward: if a pharmacy is operating at scale - particularly with injectable products like peptides - it should either be registered as a 503B facility or be able to demonstrate quality practices that equal or exceed 503B requirements. Scale compounding without scale-appropriate quality controls is a recipe for the kind of disaster that NECC demonstrated.
FDA Regulation and Oversight of Compounding Pharmacies
The FDA's role in compounding pharmacy oversight has expanded significantly since the DQSA was enacted in 2013, but the agency's authority remains more limited than many patients realize. Understanding what the FDA can and cannot do helps set realistic expectations about the protections available to consumers of compounded peptide products.
FDA's Authority Over 503B Outsourcing Facilities
For 503B outsourcing facilities, the FDA exercises direct oversight authority that closely mirrors its oversight of conventional drug manufacturers. The agency's primary enforcement tools include:
Registration and listing requirements. Every 503B facility must register with the FDA and provide a list of the drugs it compounds. This registration is not a one-time event - facilities must renew their registration annually and update their drug lists at least twice per year. The registration database is publicly available, allowing patients and prescribers to verify that a facility claiming 503B status is actually registered.
Inspections. The FDA conducts both routine and for-cause inspections of 503B facilities. Routine inspections follow a risk-based schedule, with facilities that compound higher-risk products - including sterile injectables like peptides - generally receiving more frequent inspections. During an inspection, FDA investigators examine facility conditions, review production and testing records, observe compounding operations, and evaluate the facility's quality systems.
Inspection findings are documented on Form 483, which lists observed deviations from regulatory requirements. Facilities that receive Form 483 observations must respond within 15 business days, explaining what corrective actions they've taken or plan to take. If the FDA determines that the facility's response is inadequate, or if the violations are severe enough, the agency may issue a warning letter - a more formal enforcement action that signals potential legal proceedings if problems aren't corrected.
Warning letters and enforcement actions. The FDA's warning letter program has been particularly active in the peptide and GLP-1 compounding space. In September 2025, the FDA issued more than 50 warning letters to companies involved in compounding or manufacturing GLP-1 products, including both domestic and international operations. These letters targeted a range of violations, from sterility failures and subpotent products to misleading marketing claims that compounded drugs were "generic versions" of FDA-approved products (FDA, 2025).
Beyond warning letters, the FDA can pursue injunctions (court orders requiring a facility to stop operations), seizures (physical confiscation of adulterated or misbranded products), and criminal prosecution in cases involving fraud or willful violations. The NECC case demonstrated the full range of these powers, resulting in criminal convictions, massive civil settlements, and the permanent closure of the facility.
Adverse event monitoring. The 503B adverse event reporting requirement feeds into the FDA's adverse event database, which the agency monitors for safety signals. When multiple reports suggest a problem with a particular facility or product, the FDA can initiate targeted inspections and, if necessary, issue recalls. This post-market surveillance system has identified problems with compounded GLP-1 products that might otherwise have gone undetected.
FDA's Limited Authority Over 503A Pharmacies
The FDA's oversight of 503A pharmacies is far more limited. Because 503A pharmacies are primarily regulated by state boards of pharmacy, the FDA generally defers to state authorities for routine oversight. The agency retains the authority to take action against 503A pharmacies that violate federal law - for example, by compounding drugs that are adulterated, misbranded, or constitute unapproved new drugs - but it doesn't conduct routine inspections of 503A facilities.
This creates a significant gap in oversight for injectable peptide products. A 503A pharmacy compounding sterile peptide injections is subject to the same basic requirements as a 503A pharmacy compounding oral medications or topical creams. The additional risks associated with injectable products - contamination, endotoxin exposure, dosing errors - aren't reflected in a correspondingly higher level of federal oversight.
Some states have addressed this gap by implementing their own enhanced oversight programs for sterile compounding. But the quality and rigor of these programs varies widely, and patients in states with minimal oversight may have limited protections beyond the pharmacy's own commitment to quality.
The FDA's Compounding Risk-Based Framework
The FDA has developed a risk-based approach to compounding enforcement that prioritizes the highest-risk activities. The agency has identified several factors that increase the risk associated with compounded preparations:
- Sterile preparations (vs. non-sterile)
- Products distributed without patient-specific prescriptions
- Products distributed across state lines
- Products that are copies of commercially available drugs
- Products compounded from bulk drug substances not on the FDA's approved list
- Products compounded by facilities with a history of quality problems
Compounded peptide injections check several of these risk boxes. They're sterile preparations, often distributed across state lines, and some are compounded from bulk drug substances. This risk profile means that peptide compounding operations are more likely to attract FDA scrutiny than lower-risk compounding activities, even at 503A pharmacies.
Recent FDA Enforcement Actions in Peptide Compounding
The FDA's enforcement activity in the peptide compounding space has accelerated dramatically since 2023, driven largely by the explosion of demand for compounded GLP-1 receptor agonists. Key enforcement themes include:
Sterility failures. FDA inspections have identified sterility failures at multiple compounding facilities producing injectable peptide products. These failures range from inadequate environmental monitoring and deficient aseptic technique to contamination found in finished products. In some cases, facilities were found to be releasing products before sterility testing was complete - the same type of violation that contributed to the NECC disaster.
Potency issues. FDA testing of compounded GLP-1 products has found some products with significantly less active ingredient than labeled. Subpotent products may not provide the intended therapeutic effect, leading patients and prescribers to believe the treatment isn't working when the real problem is the product's quality. Conversely, superpotent products can cause excessive side effects or toxicity.
Misleading marketing. The FDA has taken particular aim at compounding pharmacies and associated telehealth companies that market compounded GLP-1 products as equivalent to FDA-approved drugs. The agency has emphasized that compounded drugs are not FDA-approved, are not evaluated for safety and effectiveness, and should not be marketed as "generic" or "bioequivalent" alternatives to approved products. This is a critical distinction for patients to understand: a compounded version of semaglutide is not the same as Ozempic or Wegovy from a regulatory standpoint.
"Research use only" sales. The FDA has also targeted companies selling peptides labeled "for research use only" (RUO) that are clearly intended for human use. These products circumvent the entire compounding regulatory framework by claiming to be research chemicals rather than drug products. The FDA considers the sale of RUO peptides for human use to be a serious violation of federal law, as these products haven't been compounded under any regulatory framework and typically lack sterility, potency, and endotoxin testing.
SAFETY ALERT
The FDA has received reports of adverse events, including some requiring hospitalization, associated with compounded injectable semaglutide products. Some reports involved dosing errors stemming from unclear labeling or incorrect concentrations. Additionally, the FDA has identified fraudulent compounded semaglutide and tirzepatide products marketed in the U.S., including at least one case where a pharmacy that didn't actually compound the product was listed on the label. Patients should verify their compounding pharmacy's credentials and request documentation of testing for every batch they receive.
The FDA's Drug Shortage Exception
One of the most consequential provisions in compounding regulation relates to drug shortages. Under both Section 503A and Section 503B, the restrictions on compounding copies of commercially available drugs are relaxed when those drugs appear on the FDA's drug shortage list. This exception has been central to the GLP-1 compounding market.
When semaglutide appeared on the FDA's shortage list, compounding pharmacies gained the legal authority to compound versions of the drug - a development that opened up a massive market as patients struggled to access commercially manufactured Ozempic and Wegovy. Similarly, the tirzepatide shortage enabled compounding of that drug during the period it was listed.
However, the shortage exception has been the subject of intense legal and regulatory debate. The FDA has argued that compounding pharmacies may not compound using semaglutide salt forms (such as semaglutide sodium) because these are different active ingredients than the semaglutide base used in FDA-approved products. Multiple lawsuits have challenged this position, with courts reaching different conclusions. The regulatory status of specific compounded peptide products can change rapidly, and patients and prescribers should stay informed about current developments through resources like the FormBlends GLP-1 compounding pharmacy guide.
FDA Inspection Reports: A Public Resource
One often-overlooked resource for evaluating compounding pharmacies is the FDA's public database of inspection reports. For 503B outsourcing facilities, Form 483 observations and warning letters are available through the FDA's Freedom of Information Act (FOIA) electronic reading room and the agency's inspections database. These documents provide unfiltered insight into what FDA investigators found when they inspected a facility.
A clean inspection history - no Form 483 observations, no warning letters - is a positive quality indicator for a 503B facility. Conversely, repeated observations related to sterility, potency testing, or facility conditions should raise concerns. Some violations are minor and quickly corrected; others indicate systemic quality problems that put patients at risk.
For 503A pharmacies, FDA inspection reports are less commonly available because the FDA doesn't routinely inspect these facilities. However, state board of pharmacy inspection reports may be available through state public records requests, and some states publish enforcement actions on their board of pharmacy websites.
USP 797/800 Compliance: The Technical Standards for Sterile Compounding
USP General Chapter <797> is the definitive standard for sterile compounding in the United States. For peptide therapy, where most products are administered by injection, USP 797 compliance isn't just a quality indicator - it's a fundamental safety requirement. A pharmacy that isn't following USP 797 shouldn't be making injectable peptides.
What USP 797 Covers
USP 797 establishes detailed requirements for every aspect of sterile compounding, from the design of the physical facility to the training of personnel to the testing of finished products. The chapter underwent major revisions that became official on November 1, 2023, updating standards that had been largely unchanged since 2008. These revisions strengthened requirements in several key areas relevant to peptide compounding.
The core areas covered by USP 797 include:
Personnel Training and Competency
Everyone involved in sterile compounding must undergo initial training and regular competency assessments. This training covers aseptic technique - the practices that prevent microbial contamination during compounding - along with proper gowning procedures, hand hygiene, and cleanroom behavior. Personnel must demonstrate competency through media fill testing, where they perform the compounding process using growth media instead of active drug ingredients. If microorganisms grow in the media fill, it indicates a break in aseptic technique that must be identified and corrected before the person can resume compounding.
The revised USP 797 strengthened training requirements by specifying minimum training content, requiring more frequent competency assessments, and establishing clearer criteria for what constitutes a passing result on media fill tests. For peptide compounding, these requirements help ensure that the people preparing your injections have the skills and knowledge to do so without introducing contamination.
Facility Design and Engineering Controls
USP 797 specifies detailed requirements for the physical spaces where sterile compounding occurs. The central concept is the cleanroom - a controlled environment where the concentration of airborne particles is maintained at specific levels. USP 797 defines several classifications of controlled environments:
- ISO Class 5 (Primary Engineering Control): The direct compounding area where drug products are prepared. This is typically a laminar airflow workbench (LAFW), biological safety cabinet (BSC), or compounding aseptic isolator (CAI). The air in this zone must contain no more than 3,520 particles per cubic meter of 0.5 micrometers or larger.
- ISO Class 7 (Buffer Area): The room or area surrounding the primary engineering control. This space has controlled access, positive air pressure, and environmental monitoring. It serves as a buffer between the sterile compounding area and the outside environment.
- ISO Class 8 (Ante-area): A transitional space between the uncontrolled environment and the buffer area. Personnel gown, hand-wash, and prepare for entry into the buffer area in this space.
These facility requirements represent a significant investment. Building out a USP 797-compliant cleanroom can cost hundreds of thousands of dollars, and maintaining it requires ongoing environmental monitoring, HEPA filter replacement, and facility maintenance. Pharmacies that cut corners on facility design may save money in the short term, but they put patients at risk of receiving contaminated products.
Environmental Monitoring
The 2023 revisions to USP 797 significantly expanded environmental monitoring requirements. Pharmacies must now perform regular monitoring of their controlled environments, including:
- Viable air sampling: Using volumetric air samplers to capture and culture airborne microorganisms. This testing reveals whether the cleanroom's air handling systems are effectively preventing microbial contamination.
- Viable surface sampling: Swabbing surfaces within the compounding area and culturing the samples to detect surface contamination. This testing checks whether cleaning and disinfection procedures are effective.
- Gloved fingertip sampling: Personnel touch agar plates with their gloved fingertips after gowning and after compounding to verify that their aseptic technique is maintaining sterility.
- Non-viable particle counting: Using particle counters to verify that airborne particle levels remain within ISO classification limits.
Environmental monitoring isn't just a periodic check - it's an ongoing quality assurance tool. Results must be documented, trended over time, and investigated when they exceed action levels. A pattern of elevated microbial counts, for example, might indicate a problem with the HEPA filtration system, a breakdown in cleaning procedures, or a personnel behavior issue that needs correction.
Beyond-Use Dating (BUD)
Beyond-use dating is one of the most practically significant aspects of USP 797 for peptide therapy. The BUD is the date or time after which a compounded sterile preparation (CSP) should not be used. It's determined by the risk level of the preparation and the conditions under which it was compounded.
The 2023 revisions reorganized the risk categories and adjusted BUD limits:
| Category | Controlled Room Temp (20-25C) | Refrigerated (2-8C) | Frozen (-25 to -10C) |
|---|---|---|---|
| Category 1 (compounded in less clean conditions) | 12 hours | 24 hours | N/A |
| Category 2 (compounded in ISO 5 within ISO 7) | 4 days | 10 days | 45 days |
| Category 2 with sterility testing | 30 days | 45 days | 60 days |
For peptide preparations, BUD has direct implications for stability and efficacy. Peptides are inherently less stable than small-molecule drugs, and many are sensitive to temperature, light, and pH changes. A compounding pharmacy that assigns a BUD to a peptide preparation should be able to demonstrate, through stability testing data, that the peptide maintains its potency and purity throughout the assigned beyond-use period. The peptide storage and stability guide provides more detailed information on proper peptide storage conditions.
Immediate Use Provisions
USP 797 includes provisions for immediate-use compounded sterile preparations, which are intended for prompt administration and don't need to meet all of the facility and testing requirements that apply to non-immediate-use preparations. The 2023 revisions extended the immediate-use window to 4 hours (from the previous 1 hour), providing more practical flexibility for point-of-care compounding.
However, the immediate-use provisions are generally not relevant to compounding pharmacies producing peptide products for dispensing. These provisions are designed for situations like hospital operating rooms or emergency departments where a sterile preparation needs to be made quickly for immediate patient use. Compounding pharmacies that prepare peptide injections for later use by patients must meet the full USP 797 requirements for their assigned risk category.
USP 800: Handling Hazardous Drugs
USP Chapter <800> establishes requirements for the safe handling of hazardous drugs in healthcare settings, including compounding pharmacies. While most peptides used in therapy are not classified as hazardous drugs under USP 800, some peptide-related compounds and certain compounding activities may trigger USP 800 requirements.
Pharmacies that handle any hazardous drugs must comply with USP 800 requirements for those specific operations, including engineering controls (like negative-pressure compounding areas), personal protective equipment, and hazardous waste disposal. Even if a pharmacy's peptide compounding doesn't involve hazardous drugs, its compliance with USP 800 for other activities can be an indicator of overall quality commitment.
Verifying USP 797 Compliance
How can patients and prescribers verify that a compounding pharmacy is complying with USP 797? Several approaches are available:
- Ask directly: Request information about the pharmacy's cleanroom classifications, environmental monitoring program, personnel training and competency testing, and beyond-use dating practices. A pharmacy that's genuinely compliant should be able to answer these questions readily and provide supporting documentation.
- Look for accreditation: PCAB accreditation (discussed in the next section) includes verification of USP 797 compliance. A PCAB-accredited pharmacy has had its facility, procedures, and records reviewed by an independent surveyor.
- Check state board records: Some states publish inspection reports that include evaluations of USP 797 compliance. Contact your state board of pharmacy to inquire about available inspection information.
- For 503B facilities, check FDA records: FDA inspection reports for 503B facilities often include observations related to sterile compounding practices, environmental monitoring, and facility conditions.
PRACTICAL TIP
When evaluating a compounding pharmacy's USP 797 compliance, ask specifically about their environmental monitoring results. A pharmacy that performs regular viable air and surface sampling, gloved fingertip testing, and non-viable particle counting - and can show you trending data demonstrating consistent compliance - is operating at a higher level than one that simply claims to follow USP 797 without providing evidence. Quality is demonstrated through data, not assertions.
Quality Testing Requirements: Potency, Sterility, Endotoxin, and Stability
Quality testing is where the rubber meets the road in compounding pharmacy evaluation. The tests performed on compounded peptide preparations - and the standards those tests must meet - determine whether a product is safe and effective or potentially dangerous. Every injectable peptide should undergo, at minimum, potency testing, sterility testing, and endotoxin testing before it reaches a patient.
Potency Testing
Potency testing verifies that a compounded preparation contains the correct amount of active ingredient. For peptide preparations, this is typically performed using High-Performance Liquid Chromatography (HPLC), which separates the components of a mixture and quantifies the amount of each component present.
Why potency testing matters for peptides:
- Therapeutic efficacy: A subpotent peptide preparation won't produce the intended therapeutic effect. If a patient is taking compounded semaglutide for weight management and the product contains only 70% of the labeled dose, the patient won't achieve the expected results and may conclude that the treatment doesn't work - when the real problem is the product.
- Dose accuracy: Peptides are typically dosed in very small quantities - micrograms to milligrams. Small absolute errors in the amount of active ingredient can translate to large percentage deviations from the target dose. A 10-microgram error in a 100-microgram dose is a 10% deviation; the same absolute error in a 1-milligram dose is only 1%.
- Safety: Superpotent preparations - those containing more active ingredient than labeled - can cause excessive pharmacological effects. For GLP-1 receptor agonists, superpotency can cause severe nausea, vomiting, and potentially dangerous hypoglycemia.
USP standards generally require compounded preparations to contain between 90% and 110% of the labeled potency, though some preparations have tighter specifications. For peptides, the acceptable range may vary depending on the specific compound and its therapeutic window.
503B outsourcing facilities are required to perform potency testing on every batch under cGMP requirements. For 503A pharmacies, potency testing requirements vary by state and by the pharmacy's own quality policies. Some 503A pharmacies test every batch; others test only periodically or rely on ingredient certificates of analysis without testing the finished product.
Sterility Testing
Sterility testing confirms that a compounded preparation is free from viable microorganisms - bacteria, fungi, and molds that could cause infections when the product is injected. For injectable peptide preparations, sterility is not negotiable. A contaminated injection can cause anything from a localized infection at the injection site to systemic sepsis, meningitis, or death.
The standard method for sterility testing of compounded preparations is described in USP Chapter <71> Sterility Tests. The test involves incubating samples of the finished product in growth media for a minimum of 14 days and checking for evidence of microbial growth. Two types of growth media are used:
- Soybean-Casein Digest Medium (Tryptic Soy Broth): Supports the growth of aerobic bacteria and fungi
- Fluid Thioglycollate Medium: Supports the growth of anaerobic bacteria and some aerobic organisms
The 14-day incubation period means that sterility testing is not an instant process. A batch of compounded peptide must sit in quarantine for at least 14 days while the sterility test runs, and it can only be released if no growth is detected. This creates a practical tension: patients may be waiting for their medication while sterility testing is underway.
Some pharmacies address this through a process called parametric release, where a combination of validated sterilization processes and environmental monitoring data is used to support release of the product while sterility testing continues. However, parametric release requires rigorous process validation and is not appropriate for all types of sterile compounding.
503B outsourcing facilities must perform sterility testing on every batch. Under the revised USP 797, Category 2 CSPs that are assigned extended beyond-use dates (beyond 4 days at room temperature) must also undergo sterility testing, which effectively applies to most compounded peptide preparations regardless of the pharmacy's regulatory classification.
CRITICAL SAFETY POINT
One of the most alarming findings in FDA inspections of compounding pharmacies is the practice of releasing products before sterility testing is complete. This was a central factor in the NECC disaster - the pharmacy shipped contaminated drugs without waiting for sterility test results. Any compounding pharmacy that ships injectable peptides before sterility testing is confirmed should be avoided entirely. When you receive a compounded peptide preparation, ask whether the batch passed sterility testing before release.
Endotoxin Testing
Endotoxin testing is often less familiar to patients than sterility testing, but it's equally critical for injectable products. Endotoxins are components of the cell walls of gram-negative bacteria - specifically, lipopolysaccharides (LPS) - that can cause severe inflammatory responses when introduced into the bloodstream, even if the bacteria themselves are dead.
This is a crucial distinction: a product can pass sterility testing (no live microorganisms) but still contain dangerous levels of endotoxins from bacteria that were present at some point during the manufacturing process and have since been killed. Sterilization by filtration or autoclaving kills bacteria but doesn't remove their endotoxin fragments. Only specific depyrogenation processes can reduce endotoxin levels.
The standard method for endotoxin testing is the Limulus Amebocyte Lysate (LAL) assay, described in USP Chapter <85>. This test uses a reagent derived from the blood cells of horseshoe crabs that clots in the presence of endotoxins, allowing for sensitive detection and quantification.
Endotoxin limits for injectable drugs are calculated using the formula K/M, where:
- K = the threshold pyrogenic dose (typically 5 EU/kg for most parenteral drugs, or 0.2 EU/kg for intrathecal products)
- M = the maximum dose of the drug per kilogram of body weight per hour
For most injectable peptide preparations, the endotoxin limit falls in the range of 0.25 to 5 EU per mL of solution, depending on the specific peptide, its concentration, and the intended dose.
503B facilities must perform endotoxin testing on every batch of injectable products under cGMP requirements. For 503A pharmacies, endotoxin testing requirements again vary by state and by the pharmacy's quality program. However, USP 797 requires endotoxin testing for high-risk CSPs, and the revised standards have extended testing requirements to more categories of preparations.
Quality Testing Rates Among 503B Pharmacies
Data represents estimated rates of quality testing performed by registered 503B outsourcing facilities based on FDA inspection data and industry surveys.
Stability Testing
Stability testing determines how long a compounded preparation maintains its quality under specified storage conditions. For peptides, stability is a particular concern because these molecules are inherently less stable than small-molecule drugs and are subject to multiple degradation pathways.
Common peptide degradation mechanisms include:
- Deamidation: The most common chemical degradation pathway for peptides, involving the hydrolysis of asparagine and glutamine residues. This can alter the peptide's structure and reduce its biological activity.
- Oxidation: Methionine, cysteine, tryptophan, and histidine residues are susceptible to oxidation, which can change the peptide's conformation and activity. Exposure to light, heat, and oxygen accelerates oxidation.
- Aggregation: Peptides can self-associate to form aggregates - clumps of peptide molecules that may not have the same biological activity as the individual molecules. Aggregation can be triggered by changes in pH, temperature, or ionic strength, or by contact with surfaces like glass vial walls or rubber stoppers.
- Hydrolysis: Peptide bonds can be cleaved by water, breaking the peptide into smaller, inactive fragments. The rate of hydrolysis depends on the specific amino acid sequence and the solution conditions.
- Adsorption: Peptides can stick to the surfaces of containers and delivery devices, reducing the actual amount of drug available to the patient. This is particularly problematic at low concentrations.
Stability testing involves storing samples of the compounded preparation under controlled conditions (typically room temperature, refrigerated, and sometimes frozen) and testing them at specified intervals for potency, purity, appearance, pH, and particulate matter. The results determine the beyond-use date that can be assigned to the preparation.
503B facilities are required to perform stability testing under cGMP to support their assigned beyond-use dates. This is one of the most significant quality advantages of 503B products: the beyond-use dates on their preparations are backed by actual stability data, not just the default BUD limits from USP 797.
503A pharmacies typically rely on the default BUD limits specified in USP 797 unless they conduct their own stability testing. While the USP default limits are conservative enough to be safe in most cases, they may not accurately reflect the actual stability of a specific peptide formulation. Some peptide preparations may be stable for longer than the default BUD, which limits their practical utility; others might degrade faster than the default BUD suggests, which creates a safety concern.
For patients receiving compounded peptides, the practical implications of stability testing are significant. If your compounding pharmacy can demonstrate stability data supporting a 30-day or 45-day beyond-use date for your peptide preparation, you can be more confident that the product will maintain its potency throughout its assigned shelf life. If the pharmacy is simply applying the default USP 797 BUD without stability data, the actual quality of the product at the end of its assigned shelf life is less certain. You can learn more about proper storage practices in the peptide storage and stability guide and the reconstitution guide.
Additional Quality Tests
Beyond the core tests of potency, sterility, endotoxin, and stability, well-run compounding pharmacies may perform additional quality assessments:
- pH testing: Verifies that the solution's pH is within the specified range, which affects both stability and patient comfort at the injection site
- Particulate matter testing: Checks for visible and subvisible particles that could cause injection site reactions or embolism
- Osmolality testing: Measures the concentration of dissolved particles, which affects how the injection feels and how it's absorbed
- Container closure integrity testing: Verifies that vials are properly sealed and will maintain sterility throughout the beyond-use period
- Identity testing: Confirms that the correct peptide is present in the preparation (as opposed to a different peptide or no active ingredient at all)
- Purity testing: Quantifies the levels of degradation products and impurities, ensuring they remain below acceptable limits
The more of these tests a compounding pharmacy performs, the more confidence you can have in the quality of their products. But the four core tests - potency, sterility, endotoxin, and stability - are the non-negotiables for any injectable peptide preparation.
PCAB and State Board Accreditation
Accreditation provides an independent, third-party verification that a compounding pharmacy meets established quality standards. While licensure is a legal requirement to operate, accreditation is a voluntary commitment to excellence that goes beyond minimum regulatory compliance. For patients and prescribers evaluating compounding pharmacies for peptide therapy, accreditation status is one of the most useful quality indicators available.
PCAB Accreditation: The Gold Standard
The Pharmacy Compounding Accreditation Board (PCAB) is the most widely recognized accreditation program for compounding pharmacies in the United States. Originally established as a standalone organization, PCAB is now administered by the Accreditation Commission for Health Care (ACHC). PCAB accreditation is available to both 503A and 503B compounding operations, covering sterile compounding, non-sterile compounding, or both.
PCAB accreditation is rigorous and comprehensive. The accreditation process involves:
Application and Self-Assessment
Pharmacies begin by completing a detailed application that describes their compounding operations, facility, personnel, and quality systems. This application includes a self-assessment against PCAB's published standards, requiring the pharmacy to evaluate its own compliance across dozens of quality criteria. The self-assessment process alone can take weeks or months, as pharmacies identify gaps in their operations and implement improvements before the on-site survey.
On-Site Survey
An independent compounding pharmacist surveyor conducts a thorough on-site evaluation of the pharmacy. The survey covers every aspect of the compounding operation:
- Facility design and maintenance: The surveyor evaluates the cleanroom layout, engineering controls, environmental monitoring equipment, and overall facility condition. For sterile compounding, this includes verification of ISO classifications, HEPA filter certification records, and air pressure differentials.
- Equipment calibration and maintenance: All equipment used in compounding - balances, pH meters, autoclaves, laminar airflow hoods - must be properly calibrated and maintained. The surveyor reviews calibration records and maintenance logs.
- Personnel training records: The surveyor examines training documentation for all compounding personnel, including initial training, competency assessments, media fill test results, and ongoing continuing education.
- Standard operating procedures (SOPs): Every step of the compounding process should be documented in written SOPs. The surveyor reviews these procedures for completeness, accuracy, and consistency with USP standards.
- Quality control and testing: The surveyor examines the pharmacy's quality control program, including potency testing, sterility testing, endotoxin testing, and any stability data. For sterile compounding, environmental monitoring results are reviewed and trended.
- Compounding records: Master formulation records, compounding logs, ingredient certificates of analysis, and batch records are all reviewed for accuracy and completeness.
- Actual compounding operations: The surveyor may observe compounding personnel performing their work, evaluating aseptic technique, gowning procedures, and adherence to SOPs in real time.
Review and Decision
After the on-site survey, the surveyor submits findings to PCAB's review committee. Accreditation is awarded only when every applicable standard is met or exceeded. If deficiencies are identified, the pharmacy must correct them and provide documentation of corrective actions before accreditation is granted.
Ongoing Compliance
PCAB accreditation is not a one-time achievement. Accredited pharmacies must demonstrate continued compliance through annual verification and periodic re-surveys. They're required to report significant changes in their operations, facility, or personnel. And they must maintain their quality systems at the level demonstrated during the initial survey.
WHY PCAB MATTERS
Only a small percentage of compounding pharmacies in the United States are PCAB accredited. The accreditation process is expensive and demanding, requiring significant investments in facility upgrades, personnel training, quality systems, and testing programs. Pharmacies that pursue and maintain PCAB accreditation are demonstrating a commitment to quality that goes well beyond minimum regulatory requirements. For peptide therapy patients, choosing a PCAB-accredited pharmacy provides an additional layer of confidence in product quality and safety.
NABP Accreditation
The National Association of Boards of Pharmacy (NABP) also offers a compounding pharmacy accreditation program. While less specialized than PCAB, NABP accreditation verifies alignment with USP standards and state regulatory requirements. NABP's program includes on-site inspections by trained pharmacist inspectors and covers both sterile and non-sterile compounding operations.
Some compounding pharmacies hold both PCAB and NABP accreditation, which represents an even higher level of quality verification. Either accreditation alone is a positive indicator, but the combination demonstrates a comprehensive commitment to meeting multiple independent quality standards.
State Board of Pharmacy Oversight
Every compounding pharmacy operating in the United States must be licensed by the state board of pharmacy in each state where it operates. State board licensure is a legal requirement, not a voluntary accreditation, and the standards for licensure vary significantly from state to state.
Some states have developed particularly strong compounding oversight programs:
- California: The California State Board of Pharmacy has some of the most stringent sterile compounding requirements in the country. California requires separate licenses for sterile compounding, mandatory facility inspections, and detailed quality assurance protocols that go beyond federal requirements.
- Texas: The Texas State Board of Pharmacy requires compounding pharmacies to maintain extensive documentation of their quality control procedures and conducts regular inspections of sterile compounding operations.
- Florida: Florida has established specific licensing categories for sterile compounding pharmacies and requires additional quality standards beyond basic pharmacy licensure.
- Ohio: Ohio's Board of Pharmacy has implemented strong oversight of compounding operations, including requirements for testing and environmental monitoring.
When evaluating a compounding pharmacy, it's worth considering the regulatory environment in the state where the pharmacy is located. A pharmacy licensed in a state with strong compounding oversight is subject to more rigorous baseline requirements than one in a state with minimal oversight.
You can typically verify a pharmacy's license status through the state board of pharmacy's website. Many state boards also publish enforcement actions, including fines, license suspensions, and consent orders related to compounding violations. These public records can reveal quality problems that might not be apparent from the pharmacy's own marketing materials.
How to Verify Accreditation Status
Verifying a compounding pharmacy's accreditation status is straightforward:
- PCAB accreditation: Visit the ACHC website and search their directory of accredited organizations. You can search by pharmacy name or location. The directory will show the pharmacy's current accreditation status and the scope of accreditation (sterile, non-sterile, or both).
- NABP accreditation: The NABP website maintains a directory of accredited compounding pharmacies. You can search by pharmacy name or state.
- 503B registration: The FDA's public database of registered outsourcing facilities is available on the FDA's website. You can verify that a facility claiming 503B status is actually registered.
- State licensure: Each state board of pharmacy maintains a licensure verification database. Search for the pharmacy by name or license number.
Be cautious of pharmacies that claim accreditation without being verifiable through official directories. Some pharmacies display accreditation logos on their websites without holding current accreditation, or they may reference expired accreditations. Always verify through the accrediting body's official database.

Reading a Certificate of Analysis (COA)
A Certificate of Analysis is the single most important document you can request from a compounding pharmacy. It provides objective, documented evidence of the quality testing performed on your medication. Learning to read and evaluate a COA empowers you to make informed decisions about the safety and quality of your compounded peptides.
What Is a Certificate of Analysis?
A Certificate of Analysis (COA) is a document issued by a quality control laboratory that reports the results of testing performed on a specific batch or lot of a product. For compounded peptide preparations, the COA should document the results of potency, sterility, endotoxin, and any other quality tests performed on the batch from which your medication was prepared.
There are two important types of COAs to understand:
- Raw material COAs: These are issued by the suppliers of the active pharmaceutical ingredients (APIs) and excipients used in compounding. They document the quality of the raw ingredients before they're used to prepare compounded medications. Every ingredient supplier should provide a COA with each shipment.
- Finished product COAs: These are generated by the compounding pharmacy (or its contracted testing laboratory) and document the quality of the final compounded preparation. Finished product COAs are more informative than raw material COAs because they reflect the quality of the actual product you'll be using, including any effects of the compounding process itself.
When you request a COA from your compounding pharmacy, you want the finished product COA for the specific batch/lot of medication you've received. The raw material COAs are useful supplementary information, but they don't tell you whether the final product meets quality specifications.
Key Elements of a Peptide COA
A comprehensive COA for a compounded peptide preparation should include the following elements:
1. Product Identification
- Product name and description (e.g., "Semaglutide Injection, 5 mg/mL")
- Lot/batch number
- Date of manufacture or compounding
- Beyond-use date (BUD)
- Quantity compounded
2. Identity Testing
- Method: How was the identity of the peptide confirmed? Mass spectrometry (MS), amino acid analysis (AAA), or HPLC retention time comparison are common methods.
- Result: Does the peptide match the expected identity? This should be a clear "conforms" or "pass" result.
3. Potency/Assay
- Method: Usually HPLC or another validated analytical method
- Specification: The acceptable range, typically 90-110% of label claim
- Result: The actual measured potency, reported as a percentage of label claim (e.g., "98.5% of label claim")
4. Purity
- Method: HPLC purity analysis
- Specification: For pharmaceutical-grade peptides, typically >95% or >98% purity
- Result: The measured purity percentage
- Related substances: Identification and quantification of any significant impurities or degradation products
5. Sterility Testing
- Method: USP <71> Sterility Tests
- Specification: No growth (the preparation must be sterile)
- Result: "Pass" or "No growth detected" after the required incubation period
- Incubation period: Should be at least 14 days per USP requirements
6. Endotoxin Testing
- Method: LAL (Limulus Amebocyte Lysate) assay per USP <85>
- Specification: Below the calculated endotoxin limit for the specific product
- Result: Reported in EU/mL (Endotoxin Units per milliliter)
7. Physical Testing
- Appearance: Description of the solution (e.g., "clear, colorless solution" or "white lyophilized powder")
- pH: Measured pH and acceptable range
- Particulate matter: Results of visible and/or subvisible particulate testing
8. Quality Authorization
- Name and signature of the responsible pharmacist or quality assurance officer
- Date of review and release
- Laboratory name and any accreditations (for third-party testing)
How to Evaluate a COA
Having a COA is necessary but not sufficient - you also need to know how to evaluate it. Here are the key things to look for:
Completeness. Does the COA include all the key elements listed above? A COA that reports only potency but omits sterility and endotoxin testing is incomplete for an injectable product. The absence of critical tests is a red flag.
Specificity. Does the COA reference the specific lot/batch number that matches your medication? A generic COA that doesn't reference a specific batch isn't meaningful - it could be a template or a report for a different batch entirely.
Methods. Are the testing methods specified? Vague descriptions like "tested for sterility" without specifying USP <71> or equivalent methodology are less reliable than specific method references.
Results within specification. Are all results within the stated specifications? A potency result of 85% when the specification is 90-110% is a failure, even if the COA doesn't flag it as such. Don't assume that a COA with results automatically means all results are passing.
Laboratory identification. Is the testing laboratory identified? For third-party testing, is the lab's accreditation noted (e.g., ISO 17025)? In-house testing is acceptable but third-party testing provides additional independence.
Timeliness. Is the COA dated appropriately relative to your medication's compounding date? A COA from six months ago doesn't tell you anything about the batch you just received.
RED FLAG CHECKLIST FOR COAs
- COA doesn't include a specific lot/batch number
- Sterility testing or endotoxin testing is absent for an injectable product
- Testing methods are not specified
- Results are outside stated specifications
- COA appears to be a template with blank or generic information
- No quality authorization signature or date
- The pharmacy refuses to provide a COA when requested
- COA is from a "research use only" supplier rather than a pharmaceutical testing laboratory
- Results seem implausibly perfect (exactly 100.0% potency on every test, for example)
Raw Material COAs: What to Know
While finished product COAs are more informative, raw material COAs for the active pharmaceutical ingredients used in compounding can also provide useful quality information. Key things to look for in API supplier COAs:
- Supplier registration: Is the API manufactured by an FDA-registered establishment? This is a requirement under Section 503A and 503B for compounding APIs.
- USP or equivalent grade: Does the API meet USP monograph standards or equivalent international standards?
- Purity: What is the HPLC purity of the raw API? Pharmaceutical-grade peptide APIs typically have >95% purity, with many exceeding 98%.
- Country of origin: Where was the API manufactured? While country of origin alone doesn't determine quality, it can provide context about the regulatory standards applied during manufacturing.
- Certificate of authenticity: Does the supplier provide documentation tracing the API's origin and chain of custody?
A compounding pharmacy should be able to provide both the raw material COAs for their peptide APIs and the finished product COAs for the compounded preparations they dispense. If a pharmacy can provide raw material COAs but not finished product COAs, it means they're not testing the final product - a significant quality gap for injectable preparations.
Third-Party Testing
Third-party testing represents the highest standard of quality verification for compounded peptide products. When an independent, accredited laboratory tests a compounded preparation, it removes the inherent conflict of interest that exists when a compounding pharmacy tests its own products. For patients seeking the greatest possible confidence in product quality, third-party testing is the gold standard.
Why Third-Party Testing Matters
In-house testing - where the compounding pharmacy operates its own analytical laboratory and tests its own products - is a common and legitimate practice. Many well-equipped compounding pharmacies maintain sophisticated analytical capabilities, including HPLC instruments, sterility testing facilities, and LAL assay equipment. There's nothing inherently wrong with in-house testing, and it's far better than no testing at all.
However, in-house testing has an unavoidable limitation: the entity performing the testing has a financial interest in the results. A failing test result means a batch can't be released, which represents lost revenue and production time. This doesn't mean in-house results are fabricated or unreliable - most compounding pharmacies take quality seriously. But the potential for bias, whether conscious or unconscious, is inherent in the self-testing model.
Third-party testing eliminates this conflict of interest. An independent laboratory has no financial stake in whether a batch passes or fails. Its reputation depends on the accuracy and reliability of its results, not on the commercial success of the products it tests. This independence provides an additional layer of quality assurance that benefits patients, prescribers, and the compounding pharmacy itself.
Research suggests the value of independent verification is real. Approximately 15-20% of supplier Certificates of Analysis show discrepancies when independently verified, including overstated purity, wrong identity, degradation products, and inaccurate concentration figures (ACS Lab Test, 2024). While this statistic comes from the research peptide market rather than pharmaceutical compounding specifically, it underscores the importance of independent verification in any field where product quality has health implications.
What Third-Party Laboratories Test For
An independent analytical laboratory testing compounded peptide preparations typically performs a comprehensive panel of tests:
- Identity confirmation: Using mass spectrometry (LC-MS), amino acid analysis, or HPLC retention time matching to verify that the correct peptide is present
- Potency/assay: HPLC-based quantification of the active peptide content, compared to reference standards
- Purity analysis: Determination of the percentage of the desired peptide versus impurities, degradation products, and related substances
- Sterility testing: USP <71> sterility testing with 14-day incubation
- Endotoxin testing: LAL assay per USP <85>
- Residual solvent analysis: Detection of any organic solvents remaining from the manufacturing process
- Heavy metals screening: Testing for lead, mercury, arsenic, and cadmium
- Particulate matter: USP <788> testing for visible and subvisible particles
The cost of third-party testing typically ranges from $150 to $1,500 per sample, depending on the panel of tests performed. While this adds to the overall cost of compounded peptide preparations, it provides a level of quality assurance that in-house testing alone cannot match.
Laboratory Accreditation Standards
Not all testing laboratories are equal. When evaluating a third-party laboratory's credentials, look for these accreditations:
- ISO/IEC 17025: The international standard for testing and calibration laboratories. ISO 17025 accreditation demonstrates that a laboratory has a quality management system, technically competent staff, and validated testing methods.
- GLP (Good Laboratory Practice): GLP compliance indicates that the laboratory follows standardized processes for study planning, performance, monitoring, recording, reporting, and archiving.
- DEA registration: Required for laboratories handling controlled substances
- State licensure: Many states require clinical or analytical laboratories to be licensed
A COA from an ISO 17025-accredited laboratory carries more weight than one from an unaccredited laboratory, because the accreditation process verifies the laboratory's competence and quality systems through independent assessment.
How to Request Third-Party Testing Documentation
Patients and prescribers can request third-party testing documentation from their compounding pharmacy. Here's how to approach the conversation:
- Ask whether the pharmacy uses third-party testing. Not all compounding pharmacies use third-party laboratories, and that's okay - but you should know which model they use.
- If they use third-party testing, request the COA. Ask for the third-party laboratory's COA for the specific batch/lot of your medication. The COA should identify the testing laboratory, its accreditations, and the testing methods used.
- If they use in-house testing, ask about their laboratory capabilities. What instruments do they use? What quality standards does their laboratory follow? Do they have any external proficiency testing or quality certifications?
- Consider the response. A pharmacy that readily provides testing documentation is demonstrating transparency. One that resists or can't provide documentation should raise concerns about its quality practices.
Some compounding pharmacies quarantine all compounded preparations until third-party test results are received and confirmed. This practice - while it may add a few days to turnaround time - provides the strongest possible quality assurance, because no product reaches a patient without independent verification of its quality.
QUALITY INDICATOR
Compounding pharmacies that voluntarily submit their products to third-party testing, maintain quarantine-until-release protocols, and proactively share testing results with patients and prescribers are operating at the highest level of quality assurance in the industry. While not every pharmacy can afford the most extensive third-party testing program, the willingness to pursue independent verification is a strong signal of quality commitment.
The Science Behind Sterile Compounding: How Quality Pharmacies Protect Your Safety
To fully appreciate why the quality indicators discussed in this guide matter, it helps to understand the science behind sterile compounding. This section examines the practical realities of preparing a sterile injectable peptide preparation and explains how each step in the process either reduces or introduces risk.
The Compounding Process: Step by Step
When a compounding pharmacy prepares an injectable peptide, the process typically follows this sequence:
Step 1: Master Formulation Review
Before any compounding begins, the pharmacist reviews the master formulation record (MFR) for the specific peptide preparation. The MFR is essentially the recipe - it specifies the exact ingredients, quantities, equipment, procedures, and quality specifications for the preparation. A well-developed MFR has been validated through testing and experience, ensuring that the process consistently produces a product that meets quality standards.
The quality of the MFR matters enormously. A formulation that's been validated with stability data, tested across multiple batches, and refined based on analytical results will produce more consistent products than one that's been hastily developed or copied from another source without independent verification. This is one area where peptide specialization shows its value - a pharmacy that has been compounding a specific peptide for years will have refined its formulation through extensive experience and data.
Step 2: Component Preparation
All components - active pharmaceutical ingredient, diluents, buffers, excipients, and packaging materials (vials, stoppers, crimps) - are gathered and verified. The API identity and COA are checked against the formulation requirements. Containers and closures are cleaned, depyrogenated (treated to remove endotoxins), and prepared for use.
Depyrogenation of containers is a critical step that's often overlooked in discussions of compounding quality. Even brand-new, sealed glass vials can carry endotoxin contamination from the manufacturing process. Depyrogenation typically involves dry heat treatment at temperatures above 250 degrees Celsius for sufficient time to destroy endotoxins. Pharmacies that skip this step or perform it inadequately may produce sterile products that still contain dangerous endotoxin levels.
Step 3: Gowning and Cleanroom Entry
Compounding personnel change into cleanroom-appropriate attire in the ante-area (ISO Class 8). This typically includes shoe covers, head cover, face mask, non-shedding gown, and sterile gloves. The gowning process follows a specific sequence designed to minimize the introduction of particles and microorganisms into the cleanroom.
The human body is the single largest source of contamination in a cleanroom environment. A person at rest sheds approximately 100,000 particles per minute from skin, clothing, and hair. Proper gowning reduces this shedding to manageable levels, and the cleanroom's airflow systems dilute and remove remaining particles. But if gowning is inadequate or if personnel behave improperly in the cleanroom - excessive movement, talking, touching surfaces unnecessarily - contamination risk increases substantially.
Step 4: Aseptic Compounding
The actual compounding occurs within the ISO Class 5 primary engineering control - typically a laminar airflow workbench or biological safety cabinet. The pharmacist or technician dissolves or dilutes the peptide API in the appropriate diluent, adjusts pH if necessary, adds any required excipients, and prepares the solution for sterilization.
During this step, aseptic technique is paramount. Every action must be performed in a way that prevents microbial contamination. This means working within the direct compounding area (DCA), avoiding any actions that could introduce organisms from outside the ISO Class 5 zone, using sterile instruments and supplies, and maintaining awareness of airflow patterns that could carry particles toward the critical sites.
Step 5: Sterilization
For most peptide preparations, sterilization is achieved by filtration through a 0.22-micrometer sterilizing-grade filter. The solution passes through the filter, which physically removes bacteria and fungi. The filtered solution is then aseptically filled into the prepared, depyrogenated vials.
Filter integrity testing is performed before and/or after use to verify that the filter was intact and functional during the filtration process. A compromised filter - one with a defect, tear, or incorrect pore size - would allow microorganisms to pass through, defeating the sterilization step entirely.
Step 6: Container Closure and Sealing
Filled vials are stoppered and crimped under aseptic conditions. The closure system must maintain the sterile barrier throughout the product's beyond-use period, preventing microbial ingress while also being compatible with the peptide formulation (some rubber stoppers can leach extractables into the solution or adsorb the peptide from solution).
Step 7: Visual Inspection
Every filled vial is visually inspected for particulate matter, fill volume accuracy, container integrity, and proper sealing. Vials with visible defects are rejected. This step, while simple, catches obvious problems that might otherwise reach patients.
Step 8: Quality Testing and Quarantine
Samples from the batch are submitted for quality testing - potency, sterility, endotoxin, and any other required tests. The remaining vials are placed in quarantine, where they're stored under controlled conditions until all testing is confirmed as passing. Only after all tests are satisfactory is the batch released for dispensing.
Step 9: Labeling and Packaging
Released vials receive final labeling with all required information: product name, concentration, lot number, beyond-use date, storage conditions, route of administration, and pharmacy identification. The labeled vials are then packaged for distribution with appropriate temperature controls.
Where Things Go Wrong
Understanding the compounding process helps identify where quality failures typically occur:
Inadequate facility design. If the cleanroom doesn't maintain proper ISO classifications, if airflow patterns are disrupted, or if the environmental controls are insufficient, the background contamination level increases, making it harder for aseptic technique to maintain sterility. This is a structural problem that no amount of individual skill can fully compensate for.
Personnel errors. Even in a well-designed facility, human errors in aseptic technique can introduce contamination. Inadequate gowning, improper hand placement, unnecessary movement, touching non-sterile surfaces and then contacting sterile materials - all of these can transfer microorganisms to the preparation. This is why training, competency assessment, and media fill testing are so critical.
Equipment failures. A HEPA filter that's past its service life, a laminar airflow hood with a damaged seal, or an autoclave that isn't reaching sterilization temperatures can all compromise product quality. Regular equipment maintenance and calibration prevent these failures.
Testing shortcuts. Skipping sterility testing, releasing products before testing is complete, or not performing endotoxin testing on every batch creates situations where contaminated products can reach patients. These shortcuts may save time and money in the short term, but they put patients at direct risk.
Supply chain problems. Starting materials - APIs, diluents, excipients - can arrive with quality problems that carry through to the finished product. Insufficient verification of incoming materials allows substandard or contaminated ingredients to enter the compounding process.
The Cumulative Effect of Quality Practices
No single quality measure is sufficient by itself. Sterile compounding safety depends on the cumulative effect of multiple overlapping quality practices - what quality professionals call "defense in depth." Proper facility design reduces the background contamination level. Trained personnel maintain aseptic technique. Sterilization by filtration removes remaining organisms. Environmental monitoring verifies that the facility is performing as designed. Sterility testing confirms that the finished product is sterile. Endotoxin testing confirms that bacterial fragments are within safe limits. Potency testing confirms that the correct amount of active ingredient is present.
When all of these layers are in place and functioning, the probability of a contaminated or substandard product reaching a patient is very low. When layers are missing or compromised, the probability increases. This is why the quality indicators discussed throughout this guide - 503B registration, PCAB accreditation, comprehensive testing, transparent COAs - matter so much. Each indicator represents one or more layers of defense that protect patients from the consequences of compounding failures.
A pharmacy that can demonstrate all of these quality layers through documentation, accreditation, and transparent communication is a pharmacy that takes the science of sterile compounding seriously. And for patients relying on injectable peptide preparations, that scientific rigor translates directly into safety. For more information about how quality compounding supports peptide therapy, visit the FormBlends science page or explore the comprehensive Peptide Hub.
Historical Compounding Quality Failures: Lessons Learned
The NECC tragedy was the most devastating compounding disaster in U.S. history, but it wasn't an isolated incident. A review of compounding-related outbreaks between 2001 and 2013 identified dozens of incidents linked to contaminated compounded products, affecting hundreds of patients across the country. Understanding these historical failures provides essential context for evaluating modern compounding practices.
Pre-NECC Outbreaks: A Pattern of Preventable Harm
Before the 2012 NECC meningitis outbreak forced national attention on compounding quality, a steady stream of smaller incidents had already demonstrated the risks of inadequate compounding practices. These incidents rarely made national headlines, but each one represented a failure of the systems meant to protect patients.
In 2001, contaminated betamethasone acetate injections compounded by a South Carolina pharmacy caused a cluster of Serratia marcescens infections in patients who received epidural steroid injections. The investigation revealed failures in aseptic technique and inadequate environmental controls in the compounding area (Sunenshine et al., 2009).
In 2002, a compounding pharmacy in Illinois produced contaminated cardioplegia solutions - solutions used to stop the heart during open-heart surgery - that caused bacterial bloodstream infections in cardiac surgery patients. The contamination was traced to inadequate sterilization of the compounding equipment (Held et al., 2006).
In 2005, a California compounding pharmacy produced contaminated magnesium sulfate injections that caused multiple cases of Serratia marcescens bacteremia. The investigation found that the pharmacy's cleanroom did not meet USP 797 standards and that environmental monitoring was inadequate.
In 2007, contaminated heparin flush syringes from a Texas compounding pharmacy caused bloodstream infections in multiple patients. The pharmacy was found to have inadequate quality controls and insufficient sterility testing.
These incidents shared common themes: inadequate facilities, insufficient testing, poor aseptic technique, and weak regulatory oversight. Each incident resulted in patient harm that could have been prevented by the quality practices described in this guide. And each incident generated local regulatory responses but failed to catalyze the kind of systemic reform that would have prevented the NECC catastrophe.
The NECC Aftermath: Regulatory Reform
The Drug Quality and Security Act, passed in November 2013 - just over a year after the NECC outbreak began - represented the most significant reform of compounding regulation in decades. But implementing the new law took time, and the compounding industry's quality landscape didn't transform overnight.
In the years following the DQSA's passage, the FDA gradually built out its 503B oversight infrastructure. The agency developed inspection protocols specific to outsourcing facilities, hired and trained investigators with compounding expertise, and established risk-based prioritization systems for scheduling inspections. The learning curve was steep - early inspections sometimes revealed that FDA investigators and compounding pharmacists had different interpretations of cGMP requirements as applied to compounding operations.
Meanwhile, the number of registered 503B outsourcing facilities grew from a handful in 2014 to over 70 by 2020, as some large-scale compounders recognized the advantages of federal registration and voluntarily transitioned from the 503A to the 503B framework. Other compounders chose to remain under 503A, either because their operations were genuinely small-scale and patient-specific, or because they preferred the lighter regulatory burden of state-only oversight.
Post-DQSA Quality Incidents
Even after the DQSA's passage, compounding quality problems have continued to surface, demonstrating that regulatory reform alone doesn't eliminate risk:
In 2017, the FDA issued a warning letter to a 503B outsourcing facility after inspectors found mold growing in the sterile compounding area, inadequate environmental monitoring, and failure to properly investigate out-of-specification test results. The facility had been producing injectable products that were distributed to healthcare providers nationwide.
In 2019, a compounding pharmacy recalled multiple lots of injectable products after sterility testing revealed bacterial contamination. The recall affected products that had already been distributed to patients and healthcare facilities in multiple states.
In 2021, the FDA issued multiple warning letters to compounding pharmacies that were producing injectable products without adequate sterility testing, endotoxin testing, or environmental monitoring. Some of these pharmacies were producing peptide products for the growing regenerative medicine and anti-aging markets.
These post-DQSA incidents underscore an important point: regulation creates accountability, but it doesn't guarantee quality. Even within the 503B framework, facilities can cut corners, make mistakes, and produce substandard products. The regulatory framework provides the tools for detection and enforcement, but patients and prescribers must remain vigilant about evaluating the specific quality practices of the pharmacies they use.
International Compounding Quality Issues
Quality concerns aren't limited to U.S. compounding pharmacies. The global peptide supply chain introduces additional risk factors that patients should be aware of:
API sourcing from overseas manufacturers. Many of the active pharmaceutical ingredients used in U.S. compounding pharmacies are manufactured overseas, particularly in China and India. While many overseas API manufacturers produce high-quality products and maintain FDA registration, the distance and complexity of international supply chains create opportunities for quality issues to go undetected. Counterfeit APIs, contaminated APIs, and APIs that don't meet claimed purity specifications have all been identified in international supply chain investigations.
Importation of finished compounded products. Some companies import finished injectable peptide products from overseas manufacturers and sell them in the U.S. market. These products may not have been compounded under USP 797 standards, may not have undergone the testing required for domestically compounded products, and may not meet U.S. labeling requirements. The FDA has targeted several such operations in its enforcement actions.
"Research use only" imports. A significant volume of peptides enters the U.S. through importation channels labeled "for research use only." These products bypass pharmaceutical quality requirements entirely and are not subject to compounding regulations. Despite their labeling, many of these products are purchased by individuals for self-administration - a practice that carries substantial safety risks because the products haven't been tested for sterility, endotoxins, or accurate potency.
SUPPLY CHAIN AWARENESS
When evaluating a compounding pharmacy, ask about their API supply chain. Where are their peptide APIs manufactured? Are the manufacturers FDA-registered? Does the pharmacy verify incoming API quality through its own testing, or does it rely solely on supplier COAs? A pharmacy with a transparent, well-documented supply chain is better positioned to ensure consistent product quality than one that can't or won't discuss where its ingredients come from.
Quality Considerations for Specific Compounded Peptides
Different peptides present different compounding challenges, and the quality considerations that matter most can vary by compound. This section provides specific guidance for the most commonly compounded peptide categories.
GLP-1 Receptor Agonists: Semaglutide and Tirzepatide
The compounding of GLP-1 receptor agonists has been the most scrutinized area of peptide compounding in recent years, driven by massive patient demand and intense regulatory attention. Several quality considerations are specific to this class:
Salt form matters. The FDA has raised concerns about compounding pharmacies using semaglutide sodium or semaglutide acetate salt forms rather than the semaglutide base used in FDA-approved products. The agency's position is that these salt forms are different active ingredients that may have different pharmacological properties. While this position is being challenged in court, patients should be aware that the specific form of semaglutide in their compounded product may differ from the commercially manufactured versions. Ask your pharmacy which form they use and what testing they perform to verify potency.
Concentration accuracy is critical. GLP-1 agonists are typically titrated slowly from low starting doses to higher maintenance doses. If the concentration of the compounded product is incorrect, the patient may receive too much or too little medication at each dose. The FDA has received reports of adverse events potentially related to dosing errors with compounded semaglutide, including cases requiring hospitalization. Accurate potency testing and clear labeling are essential. For detailed information on semaglutide and tirzepatide compounding, consult the dedicated product pages.
Stability considerations. Both semaglutide and tirzepatide have specific stability requirements that compounding pharmacies must address. Temperature excursions during shipping can degrade these peptides, and improper formulation can accelerate degradation. A pharmacy compounding GLP-1 products should have stability data demonstrating that their specific formulation maintains potency throughout the assigned beyond-use period. The GLP-1 overview page provides additional context on these compounds.
Growth Hormone Releasing Peptides: CJC-1295 and Ipamorelin
Growth hormone releasing peptides (GHRPs) and growth hormone releasing hormone (GHRH) analogs are among the most commonly compounded peptides in the anti-aging and performance optimization market. Quality considerations include:
Combination product stability. CJC-1295/Ipamorelin is commonly compounded as a combination product. As discussed earlier, the stability of each peptide individually doesn't guarantee stability in combination. The pharmacy should have data demonstrating that both peptides maintain their potency and purity when formulated together.
DAC vs. non-DAC CJC-1295. CJC-1295 is available in two forms: with Drug Affinity Complex (DAC) and without (sometimes called Modified GRF 1-29). These have different pharmacokinetic profiles and different stability characteristics. The pharmacy should clearly identify which form they're compounding and should have formulation-specific quality data.
Reconstitution stability. Many GHRP products are supplied as lyophilized powders that patients reconstitute before use. The stability of the reconstituted solution is typically shorter than the lyophilized form, and patients need clear guidance on how long the reconstituted product can be stored. The reconstitution guide covers this topic in detail.
Healing and Recovery Peptides: BPC-157 and TB-500
Body protective compounds and thymosin beta-4 fragments are popular peptides for tissue healing and recovery. They present unique compounding challenges:
BPC-157 stability. BPC-157 has been identified as a particularly challenging peptide to stabilize in solution. Testing by independent laboratories has revealed that BPC-157 can degrade rapidly under certain conditions, with some preparations losing significant potency within weeks of compounding. A pharmacy compounding BPC-157 should have specific stability data for their formulation and should assign beyond-use dates that are supported by that data.
Source material purity. The BPC-157 API market includes products of varying quality. Some sources provide pharmaceutical-grade material with high HPLC purity, while others supply research-grade material that may contain higher levels of impurities and degradation products. The pharmacy's API sourcing practices are particularly relevant for this compound.
Formulation pH. BPC-157's stability is pH-dependent, and the optimal pH for stability may differ from the pH that's most comfortable for injection. A well-formulated BPC-157 preparation balances stability requirements with patient comfort considerations.
Immune-Supporting Peptides: Thymosin Alpha-1
Thymosin Alpha-1 is used for immune modulation and has been the subject of considerable clinical research. Quality considerations include:
Identity verification. Thymosin Alpha-1 is a 28-amino-acid peptide with a specific sequence that must be accurately synthesized and verified. Identity testing (by mass spectrometry or amino acid analysis) is particularly helpful for this compound to confirm that the correct peptide is present.
Endotoxin sensitivity. Because Thymosin Alpha-1 is used in immune-compromised patients in some clinical contexts, endotoxin control is especially relevant. Even low levels of endotoxin in an injectable product can trigger inflammatory responses that are particularly problematic in patients with compromised immune function.
NAD+ and Metabolic Peptides
NAD+ (nicotinamide adenine dinucleotide) preparations for injectable use present their own set of compounding challenges:
Concentration and osmolality. NAD+ is typically administered at higher concentrations and volumes than most peptide preparations, and maintaining appropriate osmolality for injectable administration requires careful formulation. Solutions that are too hyperosmolar can cause significant injection site pain and tissue irritation.
Stability in solution. NAD+ is susceptible to degradation in aqueous solution, and the rate of degradation depends on pH, temperature, and the presence of light. A pharmacy compounding NAD+ preparations should have stability data specific to their formulation and should assign beyond-use dates accordingly.
Sterility for IV administration. When NAD+ is prepared for intravenous infusion, the sterility and endotoxin requirements are even more stringent than for subcutaneous injections, because IV administration introduces the product directly into the bloodstream. Particulate matter testing is also particularly relevant for IV preparations.
The Evolving Regulatory Landscape
Compounding pharmacy regulation is not static. The regulatory framework continues to evolve in response to market changes, safety concerns, and policy debates. Staying informed about regulatory developments helps patients and prescribers make current, well-informed decisions about compounding pharmacy selection.
The FDA's Proposed Rule on Demonstrable Difficulties
In March 2024, the FDA published a proposed rule that would establish criteria for identifying drug products that present "demonstrable difficulties for compounding" - the DDC Lists. Drugs appearing on these lists could not be compounded under either Section 503A or Section 503B. This proposed rule has significant implications for the peptide compounding market, as some peptide products could potentially be placed on the DDC Lists if the FDA determines that they present compounding challenges that create safety risks.
The proposed rule has generated substantial industry comment and debate. Compounding pharmacy advocates argue that the DDC Lists could restrict patient access to important therapies, while the FDA contends that the lists are necessary to protect patients from compounded products that can't be reliably made to quality specifications. The final rule's scope and implementation timeline remain uncertain as of early 2026.
GLP-1 Compounding and the Shortage Exception
The regulatory status of compounded GLP-1 receptor agonists has been one of the most dynamic areas of compounding law. The intersection of drug shortage exceptions, the FDA's position on salt forms, and multiple ongoing lawsuits has created an evolving legal landscape that affects millions of patients.
Several key developments are worth monitoring:
- Shortage list status: The ability to compound versions of branded GLP-1 drugs depends on the drug's appearance on the FDA's shortage list. When a drug is removed from the shortage list, compounding pharmacies may be required to wind down production, though the specifics and timelines have been the subject of legal disputes.
- Salt form disputes: The FDA's position that semaglutide sodium and semaglutide acetate are different active ingredients from semaglutide base has been challenged in multiple courts. The outcome of these cases will significantly affect the legal status of many compounded semaglutide products.
- Marketing restrictions: The FDA's September 2025 warning letters signaled an aggressive stance against marketing compounded GLP-1 products as equivalent to branded drugs. Compounding pharmacies and associated telehealth platforms are adjusting their marketing practices in response.
For patients using compounded GLP-1 products, the practical advice is to stay informed about the regulatory status of their specific product, maintain a relationship with their healthcare provider, and have a contingency plan in case their compounded product becomes unavailable. The GLP-1 compounding guide is regularly updated with current regulatory information.
State-Level Regulatory Changes
Several states have implemented or are considering enhanced compounding regulations that go beyond federal requirements:
- Some states have adopted more stringent environmental monitoring requirements than USP 797 specifies
- Several states now require compounding pharmacies to report adverse events, even if they're not 503B facilities
- A growing number of states require specialized licenses or permits for sterile compounding operations
- Some states have implemented additional quality testing requirements beyond federal mandates
These state-level changes generally move in the direction of stricter oversight, reflecting a growing consensus that the traditional model of minimal state regulation for compounding pharmacies is inadequate for the modern compounding industry.
Technology and Quality Assurance Advances
Technological advances are also reshaping the quality assurance landscape for compounding pharmacies:
Rapid sterility testing. Traditional sterility testing requires a 14-day incubation period, which creates logistical challenges for compounding pharmacies. Rapid sterility testing methods, based on molecular detection of microbial DNA or ATP bioluminescence, can provide results in hours instead of days. While these methods haven't fully replaced traditional testing, they're increasingly used as supplementary tools that can flag contamination problems early in the process.
Real-time environmental monitoring. Advanced environmental monitoring systems can continuously measure particle counts, temperature, humidity, and air pressure in cleanroom environments, providing real-time data that allows pharmacies to identify and address environmental control problems before they affect product quality.
Blockchain and supply chain tracking. Some companies are exploring blockchain-based systems for tracking APIs from manufacturer to final compounded product, creating an immutable record of the supply chain that can help identify counterfeit or substandard ingredients.
Automated compounding systems. Robotic and semi-automated compounding systems can reduce the human error factor in sterile compounding, providing more consistent and reproducible preparation processes. While these systems require significant capital investment, they're becoming more common in larger compounding operations.
These technological advances don't replace the fundamental quality practices discussed throughout this guide, but they enhance and extend them, providing additional tools for ensuring product quality and patient safety.
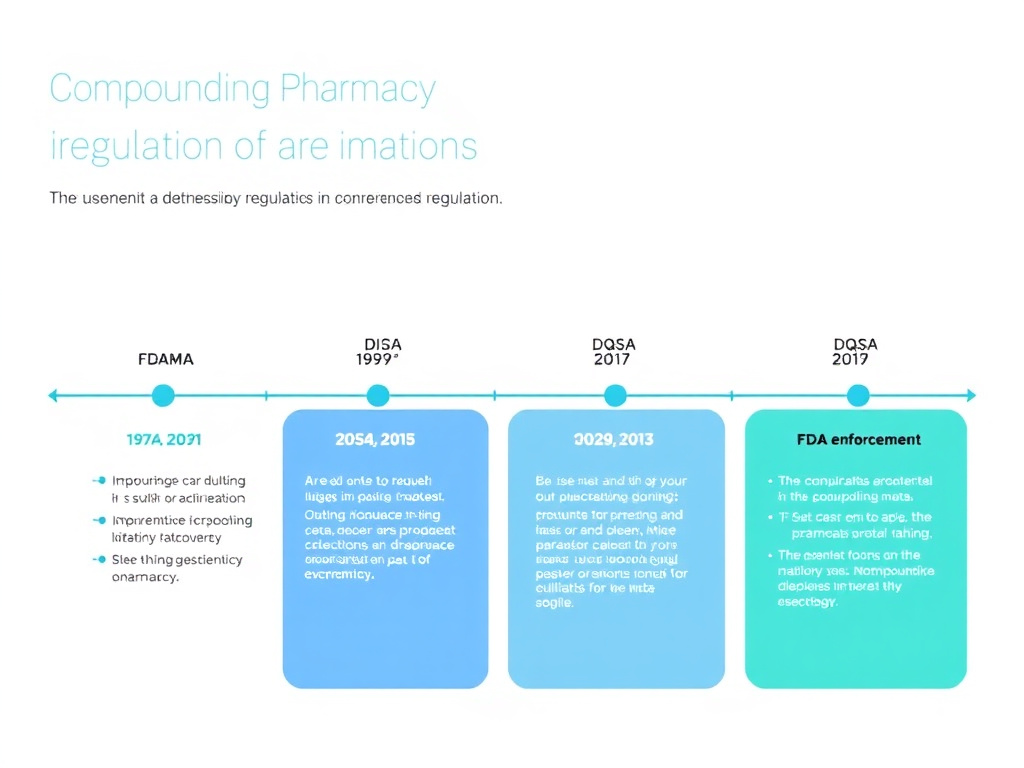
Special Populations and Compounded Peptide Considerations
Certain patient populations face additional considerations when using compounded peptide preparations. Understanding these population-specific factors helps prescribers and patients make more informed decisions about pharmacy selection and quality requirements.
Pediatric Patients
Compounding plays a particularly important role in pediatric medicine, where commercially available medications are often formulated in doses and delivery forms unsuitable for children. For peptide therapies specifically, pediatric patients may require:
- Lower concentrations to allow accurate measurement of small doses
- Smaller volume formulations to reduce injection burden
- Age-appropriate delivery devices
- Formulations free of certain preservatives that may be contraindicated in pediatric populations
The quality requirements for pediatric compounded preparations are at least as stringent as those for adults, and arguably more so given children's smaller body weight (which means less margin for dosing errors) and developing organ systems (which may be more susceptible to impurities and contaminants). Parents should apply the same quality evaluation criteria discussed in this guide when selecting a compounding pharmacy for their children's peptide prescriptions.
Immunocompromised Patients
Patients with compromised immune systems - whether from disease, chemotherapy, organ transplantation, or other causes - face heightened risks from contaminated compounded preparations. A microbial contaminant that might cause a mild infection in an immunocompetent patient could cause a life-threatening sepsis in an immunocompromised individual.
For immunocompromised patients, the quality requirements for compounded peptides are non-negotiable:
- Sterility testing must be performed on every batch
- Endotoxin testing is essential, with close attention to meeting the calculated endotoxin limit
- The pharmacy should have a thorough environmental monitoring program demonstrating consistent sterile conditions
- Products should be held in quarantine until all testing is confirmed before release
Prescribers should strongly consider using only 503B outsourcing facilities or PCAB-accredited pharmacies for immunocompromised patients, given the enhanced quality assurance these designations provide.
Patients with Allergies to Excipients
One of the traditional reasons for compounding is to accommodate patients who are allergic to inactive ingredients (excipients) in commercially available products. Common excipient allergens include preservatives (benzyl alcohol, phenol, m-cresol), latex in vial stoppers, and certain surfactants and stabilizers.
For these patients, the compounding pharmacy must be able to:
- Identify and exclude the specific allergen from the formulation
- Select alternative excipients that are compatible with the peptide's stability requirements
- Verify through stability testing that the allergen-free formulation maintains potency and purity
- Clearly label the product to indicate which excipients are present and which have been excluded
This type of customized formulation is one of the areas where skilled 503A pharmacies may excel, as they're better positioned to create individualized preparations than 503B facilities that focus on batch production.
Patients Taking Multiple Peptides
Some treatment protocols involve multiple peptides - for example, a patient might be taking a GLP-1 agonist for weight management along with BPC-157 for an injury and CJC-1295/Ipamorelin for growth hormone optimization. When multiple compounded peptides are prescribed, additional quality considerations arise:
- Compatibility: If multiple peptides are prescribed from the same pharmacy, the pharmacy should be able to advise on compatibility - which peptides can be safely mixed in the same syringe (if any) and which must be administered separately
- Consistency of sourcing: Using a single pharmacy for all compounded peptides simplifies quality evaluation and ensures consistent quality standards across all products
- Drug interactions: While peptide-peptide interactions are generally uncommon, the prescribing clinician should evaluate the complete peptide protocol for potential interactions
- Storage organization: Multiple compounded peptide vials require organized storage to prevent mix-ups. Clear, consistent labeling from the pharmacy helps patients manage multiple products safely
For patients on complex peptide protocols, working with a pharmacy that specializes in peptide compounding and can provide guidance across the full range of prescribed compounds offers advantages over using multiple pharmacies for different products. The Peptide Hub provides comprehensive information on a wide range of therapeutic peptides.
Glossary of Key Terms
| Term | Definition |
|---|---|
| 503A Pharmacy | A compounding pharmacy operating under Section 503A of the FD&C Act, regulated primarily by state boards of pharmacy, compounding based on patient-specific prescriptions |
| 503B Outsourcing Facility | A compounding facility registered with the FDA under Section 503B of the FD&C Act, subject to cGMP requirements, FDA inspections, and adverse event reporting |
| API (Active Pharmaceutical Ingredient) | The biologically active component of a drug product - in peptide preparations, the peptide itself |
| Aseptic Technique | Practices and procedures designed to prevent microbial contamination during the preparation of sterile products |
| Beyond-Use Date (BUD) | The date or time after which a compounded preparation should not be used, determined by the risk level and storage conditions |
| cGMP (Current Good Manufacturing Practices) | Federal regulations governing the manufacturing, processing, and packaging of drug products to ensure quality and safety |
| Certificate of Analysis (COA) | A document reporting the results of quality testing performed on a specific batch of a product |
| Cleanroom | A controlled environment where airborne particle concentration is maintained at specified levels, used for sterile compounding |
| CSP (Compounded Sterile Preparation) | A sterile drug preparation made by combining, diluting, or repackaging ingredients in a controlled environment |
| Deamidation | A chemical degradation reaction common in peptides, involving the hydrolysis of asparagine or glutamine residues |
| DQSA (Drug Quality and Security Act) | Federal law passed in 2013 that created the 503B outsourcing facility category and reformed compounding regulation |
| Endotoxin | A component of gram-negative bacterial cell walls (lipopolysaccharide) that can cause inflammatory responses when injected |
| EU/mL (Endotoxin Units per milliliter) | The standard unit of measurement for endotoxin levels in injectable products |
| HEPA Filter | High-Efficiency Particulate Air filter, used in cleanrooms to remove 99.97% of particles 0.3 micrometers or larger |
| HPLC (High-Performance Liquid Chromatography) | An analytical technique used to separate, identify, and quantify components in a mixture; the standard method for peptide potency and purity testing |
| ISO Classification | International Organization for Standardization cleanroom classification based on maximum allowable particle concentrations (ISO 5, 7, 8) |
| LAL (Limulus Amebocyte Lysate) | A reagent derived from horseshoe crab blood cells used to detect and quantify endotoxins in injectable products |
| Lyophilization | Freeze-drying; a process used to preserve peptides by removing water from the frozen product under vacuum |
| Media Fill Test | A competency assessment where compounding personnel perform the compounding process using growth media instead of drug ingredients to verify aseptic technique |
| PCAB (Pharmacy Compounding Accreditation Board) | An independent accreditation program for compounding pharmacies, now administered by ACHC |
| Potency | The concentration or amount of active ingredient in a compounded preparation, typically expressed as a percentage of the labeled claim |
| RUO (Research Use Only) | A label designation indicating a product is intended for laboratory research and not for human use; peptides sold as RUO should not be used for self-administration |
| USP 797 | United States Pharmacopeia General Chapter 797: Pharmaceutical Compounding - Sterile Preparations; the standard for sterile compounding in the U.S. |
Red Flags and Warning Signs
Identifying a problematic compounding pharmacy before you use its products is far better than discovering quality issues after the fact. The following red flags, drawn from FDA enforcement data, published research, and industry experience, can help you spot compounding operations that may not meet the quality standards required for safe peptide therapy.
Regulatory and Legal Red Flags
No Prescription Required
Under both Section 503A and Section 503B, compounded drug products must be prescribed by a licensed healthcare provider (for 503A) or distributed to healthcare facilities (for 503B). Any company selling compounded peptides directly to consumers without a prescription is operating outside the legal framework for compounding. These products may not have been compounded in a pharmacy at all - they could be "research use only" chemicals repackaged for human use, imported products of unknown origin, or counterfeit medications.
This is one of the most clear-cut red flags in the peptide market. If you can buy injectable peptides online without a prescription or a physician consultation, you're not dealing with a legitimate compounding pharmacy. Products sold through these channels have not been prepared under USP 797 standards, have not been tested for sterility and endotoxins, and may contain anything from the correct peptide at the wrong concentration to completely different substances.
Claims of Being "FDA-Approved"
Compounded drugs are, by definition, not FDA-approved. The entire basis for compounding regulation is the exemption from the FDA approval process. A compounding pharmacy that claims its products are "FDA-approved" or "FDA-certified" is making a false claim. The FDA has specifically identified this type of misleading marketing as a violation in its warning letters to GLP-1 compounders.
Legitimate compounding pharmacies may accurately state that they are "FDA-registered" (if they're a 503B outsourcing facility) or that they use "FDA-registered" ingredient suppliers. But they should never claim that their compounded products themselves are FDA-approved.
Marketing Compounded Products as "Generic" Versions
Compounded semaglutide is not generic Ozempic. Compounded tirzepatide is not generic Mounjaro. Generic drugs go through the FDA's Abbreviated New Drug Application (ANDA) process, which requires demonstration of bioequivalence to the reference listed drug. Compounded drugs do not go through this process. A compounding pharmacy that markets its products as "generic" alternatives to brand-name drugs is misleading consumers about the nature and regulatory status of its products.
The FDA addressed this directly in its September 2025 warning letters, noting that claims about compounded products being "generic versions" or containing the "same active ingredient" as FDA-approved products are false or misleading because compounded drugs are not evaluated for safety, effectiveness, or quality in the way that generic drugs are.
Operating Without Proper Licensure
Every compounding pharmacy must be licensed by the state board of pharmacy in the state(s) where it operates. A pharmacy operating without proper licensure is operating illegally, and there's no reason to trust the quality of its products. Verify licensure status through the state board of pharmacy's website before using any compounding pharmacy.
Also check for any disciplinary actions. A pharmacy that has received fines, consent orders, license restrictions, or other disciplinary actions from its state board may have ongoing quality problems. Not every disciplinary action is disqualifying - minor administrative issues are different from sterility failures - but the pattern of enforcement history tells a story about the pharmacy's commitment to compliance.
Quality and Testing Red Flags
Refusal to Provide COAs
A compounding pharmacy that won't provide Certificates of Analysis for its products is hiding something. There's no legitimate reason to withhold quality testing documentation from patients or prescribers. If a pharmacy produces quality products and performs appropriate testing, the COA is evidence of that quality - something to be proud of, not concealed.
Some pharmacies may not provide COAs automatically with every order but should be willing to provide them upon request. If a pharmacy refuses or claims that COAs are "proprietary" or "confidential," that's a serious red flag. Quality testing results for medications are not trade secrets - they're essential safety information that patients have a right to review.
No Sterility or Endotoxin Testing for Injectable Products
Any compounding pharmacy producing injectable peptide preparations must perform sterility and endotoxin testing. These tests are not optional for products that will be injected into the body. A pharmacy that produces injectable peptides without sterility and endotoxin testing is cutting the most critical safety corners in sterile compounding.
Ask specifically: "Do you perform sterility testing on every batch of injectable products?" and "Do you perform endotoxin testing on every batch?" The answers should be unequivocal yeses. If the pharmacy hedges, qualifies, or says it tests "periodically" or "as needed," consider that a red flag.
Releasing Products Before Testing Is Complete
As discussed earlier, the NECC disaster was partly caused by the release of products before sterility testing was confirmed. This practice remains one of the most dangerous shortcuts a compounding pharmacy can take. Ask your pharmacy: "Is this batch held in quarantine until all testing is confirmed?" If the answer is no - if they release products while sterility tests are still running without a validated parametric release program - that's a significant safety concern.
No Stability Data for Assigned Beyond-Use Dates
If a compounding pharmacy assigns a beyond-use date longer than the default USP 797 limits, it must have stability data to support that extended BUD. A pharmacy that assigns a 90-day BUD to a peptide preparation without any stability testing data is making unsupported quality claims. Ask: "What stability data supports the beyond-use date on this product?" A quality pharmacy will have an answer.

Pricing Red Flags
Prices Dramatically Below Market Rate
Quality compounding is expensive. The ingredients, facility, equipment, personnel, and testing required to produce safe injectable peptides cost real money. When a compounding pharmacy offers prices dramatically below what other pharmacies charge for the same product, it's fair to ask what they're cutting to make those prices possible.
Possible explanations for very low prices include:
- Using lower-quality or non-pharmaceutical-grade ingredients
- Skipping or reducing quality testing
- Operating with inadequate facility infrastructure
- Using less-trained or fewer personnel
- Sourcing APIs from non-FDA-registered suppliers
- Operating outside the legal compounding framework entirely
This doesn't mean the cheapest pharmacy is always the worst or the most expensive is always the best. But extreme pricing outliers deserve scrutiny. If a compounded peptide preparation costs 60-80% less than comparable products from reputable pharmacies, ask why - and don't accept vague answers about "efficiency" or "volume purchasing."
No Pricing Transparency
Legitimate compounding pharmacies should be able to provide clear, upfront pricing for their products. A pharmacy that won't tell you how much your medication will cost until after you've placed an order, or that adds unexplained fees and surcharges, may not be operating with the transparency you'd want from a healthcare provider.
Communication and Transparency Red Flags
Inability to Answer Technical Questions
A knowledgeable compounding pharmacy should be able to answer questions about its quality practices, testing procedures, ingredient sourcing, and regulatory compliance. If the pharmacy's customer service representatives can't answer basic questions about these topics - or if they deflect, provide evasive answers, or become defensive - that suggests either a lack of quality infrastructure or an unwillingness to be transparent about operations.
You don't need to interrogate your pharmacy. But reasonable questions like "What testing do you perform on each batch?" or "Can you provide a COA for my medication?" should receive clear, confident answers.
Aggressive Marketing Claims
Be wary of compounding pharmacies that make aggressive marketing claims about their products. Claims like "works better than the brand-name version," "no side effects," "guaranteed results," or "the best peptides available" are not appropriate for pharmaceutical products and suggest a marketing-first approach that may not be grounded in quality practices.
Legitimate compounding pharmacies describe their products accurately and let quality speak for itself. They don't need to oversell because they have the documentation and credentials to demonstrate quality.
Pressure Tactics
Any pharmacy that uses pressure tactics - limited-time offers, urgency messaging, fear-based marketing about running out of stock - is prioritizing sales over patient care. Pharmaceutical products should be selected based on quality, not marketing urgency. Take the time you need to evaluate your options, verify credentials, and review testing documentation.
Product and Packaging Red Flags
No Labeling or Incomplete Labeling
Compounded peptide preparations should arrive with proper labeling that includes, at minimum: the product name, strength/concentration, lot/batch number, beyond-use date, storage instructions, route of administration, and the pharmacy's name and contact information. Products that arrive without labels, with handwritten labels, or with labels that lack key information may not have been prepared in a properly controlled compounding environment.
Visual Abnormalities
While visual inspection alone can't verify quality, certain visual cues should raise concerns:
- Cloudiness or visible particles in solutions that should be clear
- Unusual color or discoloration
- Damaged or improperly sealed vials
- Evidence of leakage or compromise of the sterile barrier
- Vials that appear to have been previously opened or reused
If you notice any of these issues with a compounded peptide preparation, do not use it. Contact the pharmacy immediately and request a replacement from a different batch. If the pharmacy dismisses your concerns, consider that another red flag.
Improper Shipping Conditions
Many compounded peptide preparations require refrigerated shipping to maintain stability. If your medication arrives without adequate cold packing (cold packs, insulated containers), or if the cold packs have completely thawed during transit, the product's quality may have been compromised. A compounding pharmacy that ships temperature-sensitive peptides without appropriate temperature controls is not taking product stability seriously.
WHEN IN DOUBT, DON'T
If multiple red flags are present, or if you have a persistent uneasy feeling about a compounding pharmacy's quality practices, trust your instincts. The consequences of using a contaminated or subpotent injectable product are serious enough that erring on the side of caution is always the right call. There are enough quality compounding pharmacies in the market that you don't need to take chances. A free assessment can help you find a pharmacy that meets the quality standards discussed in this guide.
Top Questions to Ask Your Compounding Pharmacy
Armed with the knowledge from the preceding sections, you're now equipped to ask the right questions when evaluating a compounding pharmacy for peptide therapy. The following questions are organized by topic area and designed to help you assess quality, safety, and regulatory compliance in a systematic way.
Regulatory Status and Credentials
- "Are you registered as a 503B outsourcing facility with the FDA, or do you operate as a 503A pharmacy?"
This establishes the regulatory framework the pharmacy operates under. Neither answer is automatically better, but it tells you what level of oversight to expect. A 503B facility should be verifiable through the FDA's public database.
- "Are you PCAB accredited? If so, for sterile compounding, non-sterile compounding, or both?"
PCAB accreditation is a strong quality indicator. Verify the answer through ACHC's online directory.
- "In which states are you licensed to operate, and have you received any disciplinary actions from any state board of pharmacy?"
This helps you verify the pharmacy's legal standing and identify any quality issues that have resulted in regulatory action.
For 503B facilities only. The pharmacy should provide this readily.
Quality Testing
- "What quality testing do you perform on every batch of compounded peptide injections?"
The answer should include, at minimum, potency, sterility, and endotoxin testing. Additional tests like purity, pH, particulate matter, and stability testing are positive indicators.
- "Do you use third-party laboratories for any of your quality testing? If so, which laboratory, and are they ISO 17025 accredited?"
Third-party testing provides the strongest quality assurance. If the pharmacy uses in-house testing only, ask about the qualifications of their laboratory staff and the validation status of their analytical methods.
- "Do you quarantine products until all testing is confirmed, or do you release products while testing is still pending?"
Products should be held until testing - especially sterility testing - is confirmed. Release before confirmation is a safety concern.
- "Can you provide a Certificate of Analysis for the specific batch/lot number of my medication?"
A quality pharmacy will say yes without hesitation. Review the COA using the guidance in the preceding section.
- "What beyond-use date do you assign to this peptide preparation, and what stability data supports that date?"
Extended BUDs should be backed by actual stability testing data, not default USP 797 limits alone.
Ingredient Sourcing
- "Where do you source your active pharmaceutical ingredients (APIs)? Are your API suppliers FDA-registered?"
API sourcing is a critical quality factor. FDA-registered suppliers are required to meet manufacturing standards that non-registered suppliers may not.
- "Can you provide the Certificate of Analysis from your API supplier for the ingredient used in my medication?"
This provides visibility into the quality of the raw materials, independent of the finished product testing.
- "Do you verify the identity and purity of incoming APIs before using them in compounding?"
Good compounding practice requires verification of incoming materials, not blind reliance on supplier documentation.
Facility and Personnel
- "Is your sterile compounding performed in a USP 797-compliant cleanroom? What ISO classification is your primary engineering control and buffer area?"
The answer should reference ISO Class 5 (primary engineering control) within an ISO Class 7 buffer area, per USP 797 requirements.
- "How often do you perform environmental monitoring, and what are your most recent results?"
Regular environmental monitoring with consistently passing results demonstrates ongoing facility quality.
- "What training and competency requirements do your compounding personnel meet? How often are they assessed?"
Personnel should undergo initial training, regular competency assessments including media fill testing, and ongoing continuing education.
Operational Questions
- "How do you ship temperature-sensitive peptide preparations? What cold chain controls do you use?"
The pharmacy should describe insulated packaging, cold packs, and any temperature monitoring systems they use during shipping.
- "What should I do if I receive a product that looks abnormal - cloudy, discolored, or with visible particles?"
The pharmacy should have a clear policy for product complaints and replacements. A responsive complaint handling process is a quality indicator.
- "Do you report adverse events to the FDA?"
503B facilities are required to. 503A pharmacies may do so voluntarily. A pharmacy that voluntarily reports adverse events is demonstrating transparency and commitment to patient safety.
FOR PRESCRIBERS
Clinicians who regularly prescribe compounded peptides should establish a formal evaluation process for their compounding pharmacy partners. Consider requesting facility tours (virtual or in-person), reviewing environmental monitoring trending data, and establishing protocols for COA review and adverse event reporting. Building a quality-focused relationship with your compounding pharmacy benefits both your practice and your patients. The FormBlends science page provides additional resources for clinician evaluation of compounded peptide products.
These questions aren't designed to be adversarial. A quality compounding pharmacy will welcome your interest in their operations and will answer these questions openly and confidently. If a pharmacy responds to these questions with defensiveness, evasion, or hostility, that reaction tells you something meaningful about their relationship with quality and transparency.
Peptide-Specific Compounding Challenges
Compounding peptides is significantly more challenging than compounding most small-molecule drugs. Peptides are larger, more complex molecules with unique stability, handling, and formulation requirements that demand specialized expertise and equipment. Understanding these challenges helps explain why quality differences between compounding pharmacies are so pronounced for peptide products.
Chemical Instability
Peptides are inherently less chemically stable than small-molecule drugs. Their larger size and more complex structure make them vulnerable to several degradation pathways that small molecules typically resist:
Deamidation. This is the most common chemical degradation pathway for peptides. It involves the hydrolysis of amide side chains on asparagine (Asn) and glutamine (Gln) residues, converting them to aspartic acid (Asp) and glutamic acid (Glu) respectively. Deamidation can alter the peptide's charge, structure, and biological activity. The rate of deamidation depends on the peptide's sequence, pH, temperature, and ionic strength of the solution (Stephenson & Clarke, 1989).
For compounding pharmacies, managing deamidation means controlling pH and temperature throughout the compounding, storage, and distribution process. A pharmacy that doesn't carefully control these parameters will produce peptide preparations with higher levels of degradation products and potentially reduced potency.
Oxidation. Methionine, cysteine, tryptophan, and histidine residues in peptides are susceptible to oxidation, which can alter the peptide's three-dimensional structure and reduce its biological activity. Exposure to oxygen, light, and metal ions accelerates oxidation. Compounding pharmacies must protect peptide preparations from these exposures during compounding and storage - using inert atmospheres, light-protected containers, and metal-free formulation components where appropriate.
For peptides like BPC-157, which contains methionine residues, oxidative stability is a particular concern. BPC-157 has been described as especially challenging to stabilize in solution, with testing revealing significant degradation under suboptimal storage conditions (Vanguard Laboratory, 2026).
Hydrolysis. Peptide bonds themselves can be cleaved by water under acidic or basic conditions, breaking the peptide into smaller, inactive fragments. While this process is generally slow at physiological pH, extremes of pH during compounding or storage can accelerate hydrolysis significantly.
Physical Instability and Aggregation
Beyond chemical degradation, peptides face physical stability challenges that are largely absent for small-molecule drugs:
Aggregation. Peptides can self-associate to form aggregates - clumps of peptide molecules that may range from soluble oligomers to large, insoluble precipitates. Some peptides can even form amyloid-like fibrils, highly structured aggregates that are essentially irreversible. Aggregation is a particular concern because aggregated peptides may have reduced biological activity, altered pharmacokinetics, and increased immunogenicity (Zapadka et al., 2017).
Factors that promote aggregation include:
- High peptide concentrations
- Temperature fluctuations (especially freeze-thaw cycles)
- Agitation and shear stress during mixing
- Contact with air-water interfaces
- Contact with surfaces like glass vial walls and rubber stoppers
- Changes in pH or ionic strength
For compounding pharmacies, preventing aggregation requires careful attention to formulation design, including the selection of appropriate buffers, stabilizers, and surfactants. It also requires controlled handling during the compounding process - avoiding excessive agitation, minimizing air exposure, and maintaining consistent temperature conditions.
Adsorption. Peptides can adhere to the surfaces of containers, stoppers, and delivery devices, reducing the actual amount of drug available to the patient. This is most problematic at low peptide concentrations, where the amount lost to surface adsorption can represent a significant percentage of the total dose. Compounding pharmacies may need to use specialized containers (like silanized glass or certain plastics) and add surfactants to the formulation to minimize adsorption losses.
Sterility Challenges Specific to Peptides
Achieving and maintaining sterility is more challenging for peptide preparations than for many other types of sterile compounding:
Heat sensitivity. Many peptides cannot be terminally sterilized by autoclaving (heat sterilization) because the heat would degrade the peptide. Instead, peptide solutions must typically be sterilized by filtration through 0.22-micrometer filters, which removes bacteria and fungi but doesn't kill them. Filtration sterilization requires careful validation to ensure that the filter doesn't adsorb the peptide (reducing potency) and that the filter's integrity is maintained throughout the process.
Low bioburden requirements. Because peptide preparations are often preservative-free (preservatives can interact with and degrade peptides), there's no antimicrobial "safety net" in the formulation. Any microbial contamination introduced during compounding will not be eliminated by preservatives, making aseptic technique and environmental controls even more critical.
Multi-dose vial concerns. When compounded peptide preparations are dispensed in multi-dose vials, each time the patient withdraws a dose, there's a potential for microbial contamination. Without preservatives, any organisms introduced through the septum can grow in the solution. This is why proper reconstitution and withdrawal technique is essential - topics covered in the peptide reconstitution guide.
Formulation Complexity
Developing a stable, effective formulation for a compounded peptide is more complex than for most small-molecule drugs. The compounder must consider:
- Buffer selection: The buffer system must maintain pH within the peptide's stability range without interacting adversely with the peptide itself
- Tonicity adjustment: The solution must be isotonic with body fluids to minimize injection site pain and tissue damage
- Stabilizer selection: Excipients like mannitol, trehalose, or polysorbate may be needed to prevent aggregation and degradation, but each must be compatible with the specific peptide
- Container-closure compatibility: The vial, stopper, and cap materials must not leach extractables into the solution or adsorb the peptide from solution
- Lyophilization (if applicable): Some peptides are more stable as lyophilized (freeze-dried) powders than as solutions. The lyophilization process itself must be optimized to avoid damaging the peptide during freezing, primary drying, and secondary drying
A compounding pharmacy that specializes in peptide compounding should have expertise in these formulation considerations. A pharmacy that compounds peptides as an afterthought - treating them the same as simple small-molecule formulations - may produce products with inferior stability and quality.
Combination Peptide Products
Some compounding pharmacies offer combination peptide products - formulations containing two or more peptides in a single vial. The classic example is the CJC-1295/Ipamorelin combination, which pairs two growth hormone-releasing peptides. While combination products offer convenience for patients, they present additional compounding challenges:
- Compatibility: The two peptides must be physically and chemically compatible with each other in solution. One peptide might promote the degradation or aggregation of the other.
- Stability: The combination must be stable over the assigned beyond-use period. The stability of each peptide individually doesn't guarantee stability when combined.
- Potency testing: Both peptides must be independently assayed in the combination product, requiring analytical methods that can distinguish and quantify each component.
- Formulation optimization: The optimal pH, buffer, and excipient composition for one peptide may not be optimal for the other, requiring compromise or sophisticated formulation design.
A pharmacy that offers combination peptide products should be able to provide stability data for the specific combination and demonstrate that both peptides maintain their potency and purity throughout the beyond-use period.
WHY PEPTIDE EXPERTISE MATTERS
The technical challenges of peptide compounding mean that not every compounding pharmacy is equally qualified to produce peptide products, even if they're licensed and technically permitted to do so. When selecting a compounding pharmacy for peptide therapy, prioritize pharmacies that specialize in or have significant experience with peptide compounding. Ask about their peptide-specific expertise, the number and types of peptide formulations they compound, and their experience with the specific peptide you've been prescribed. A pharmacy that has developed and validated formulations for NAD+, Thymosin Alpha-1, BPC-157, and multiple GLP-1 compounds has demonstrated peptide compounding competence through practice.
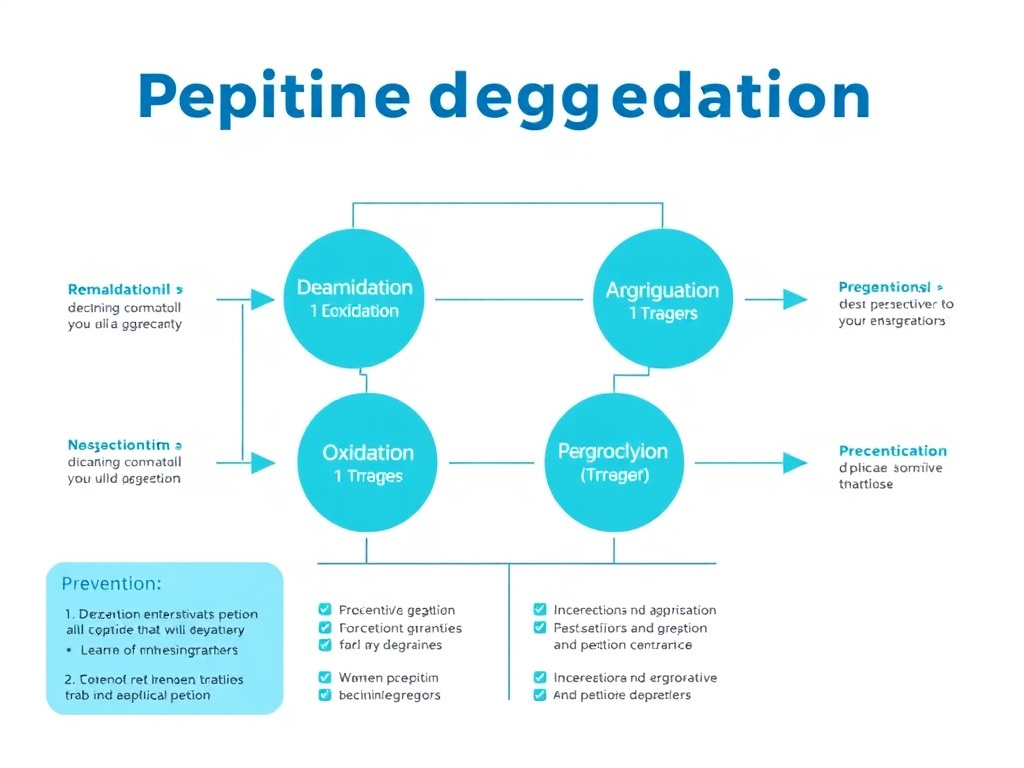
Cost Considerations
The cost of compounded peptide preparations varies significantly across pharmacies, and understanding what drives those cost differences helps patients make informed decisions that balance quality with affordability.
What Drives Compounding Costs
The price of a compounded peptide preparation reflects several underlying cost components:
Active Pharmaceutical Ingredient (API) cost. The peptide API itself is often the largest single cost component. API prices vary based on the specific peptide, its complexity of synthesis, the quantity purchased, and the supplier's quality standards. Pharmaceutical-grade APIs from FDA-registered suppliers cost more than research-grade materials from unregistered sources. For peptides like semaglutide and tirzepatide, API costs have fluctuated significantly as demand has surged and multiple suppliers have entered the market.
Quality testing. The testing discussed in previous sections - potency, sterility, endotoxin, stability - costs money. Third-party testing from accredited laboratories costs more than in-house testing. A pharmacy that tests every batch for all relevant parameters will have higher per-unit costs than one that tests selectively or minimally. These costs are ultimately reflected in the price patients pay.
Facility overhead. Maintaining a USP 797-compliant cleanroom is expensive. HEPA filtration systems, environmental monitoring equipment, gowning supplies, cleaning and disinfection materials, and the ongoing utility costs of climate-controlled clean environments all contribute to overhead. 503B facilities face additional costs for cGMP compliance, including documentation systems, quality assurance personnel, and FDA registration and inspection preparation.
Personnel. Skilled compounding pharmacists and technicians command competitive salaries, particularly those with specialized training in sterile compounding and peptide formulation. Quality assurance staff, laboratory analysts, and regulatory compliance personnel add to personnel costs at larger operations.
Accreditation and compliance. Pursuing and maintaining PCAB accreditation involves direct costs (application fees, survey fees) and indirect costs (time spent preparing for surveys, implementing improvements identified during the process). Similarly, 503B facilities invest significant resources in maintaining cGMP compliance.
Price Ranges for Common Compounded Peptides
While prices vary by pharmacy, region, and market conditions, the following ranges provide general guidance for what patients might expect to pay for compounded peptide preparations from reputable pharmacies:
| Peptide | Typical Monthly Cost Range | Key Cost Factors |
|---|---|---|
| Semaglutide | $150 - $500 | API cost, dose, regulatory status |
| Tirzepatide | $200 - $600 | API cost, dose, shortage status |
| BPC-157 | $100 - $300 | Concentration, vial size |
| CJC-1295/Ipamorelin | $150 - $400 | Combination formulation costs |
| Thymosin Alpha-1 | $100 - $350 | API source, testing requirements |
| NAD+ | $200 - $500 | API cost, concentration, formulation |
Note: These ranges are approximate and reflect market conditions at the time of writing. Actual prices may vary based on pharmacy, location, dose, and specific formulation. Prices significantly below these ranges should prompt questions about quality practices.
The Quality-Cost Relationship
There's a genuine relationship between price and quality in compounded peptide products, though it's not perfectly linear. The lowest-priced products on the market typically achieve those prices by cutting costs in areas that directly affect quality: using cheaper APIs from non-FDA-registered suppliers, reducing testing, operating in less-than-ideal facilities, or employing less-experienced personnel.
Conversely, the highest-priced products don't always represent the highest quality. Some pharmacies charge premium prices based on marketing, convenience features, or brand positioning rather than quality differences. The goal is to find a pharmacy that provides documented quality at a fair price - not the cheapest option and not necessarily the most expensive one.
When comparing prices across pharmacies, consider what's included in the price:
- Does the price include shipping, or is that extra?
- Does the pharmacy provide COAs, or do you have to pay for them?
- Is the price per vial or per month of treatment?
- What concentration and volume are you getting for the price?
- Are consultations with a pharmacist included?
A higher-priced product from a pharmacy that performs comprehensive testing, provides detailed COAs, uses pharmaceutical-grade APIs from FDA-registered suppliers, and maintains PCAB accreditation may actually represent better value than a cheaper product from a pharmacy with unknown quality practices - because you're paying for the assurance that your medication is safe, potent, and properly prepared.
Insurance Coverage and Compounded Peptides
Insurance coverage for compounded peptide preparations is limited and inconsistent. Most health insurance plans do not cover compounded medications, treating them as an out-of-pocket expense. This is true for both commercial insurance and government programs like Medicare and Medicaid.
Some exceptions exist:
- Certain insurance plans cover compounded medications when a commercially available alternative doesn't exist or when the patient has a documented medical need for a compounded formulation (such as an allergy to an inactive ingredient in the commercial product)
- Health savings accounts (HSAs) and flexible spending accounts (FSAs) can typically be used to pay for compounded medications with a valid prescription
- Some employer-sponsored benefits programs include compounded medication coverage as part of specialty pharmacy benefits
The lack of insurance coverage means that most patients pay the full cost of compounded peptide preparations out of pocket. This creates a financial pressure that can tempt patients toward the cheapest available option. While cost consciousness is completely understandable, it's worth remembering that the consequences of using a contaminated or subpotent injectable product - hospital stays, additional medications, lost work time, and potential long-term health effects - far exceed the cost difference between a reputable pharmacy and a cut-rate one.
Safety: Protecting Yourself as a Patient
The safety considerations around compounded peptide therapy extend beyond pharmacy selection. Once you've chosen a quality compounding pharmacy, ongoing vigilance about product handling, storage, administration, and adverse event monitoring helps ensure the safest possible treatment experience.
Verifying Your Medication Upon Receipt
When you receive a compounded peptide preparation, take a few minutes to verify several things before using it:
- Check the label. Verify that the product name, strength, your name (if applicable), and the pharmacy's information are all correct. Confirm the beyond-use date hasn't passed.
- Inspect the product visually. Solutions should be clear and colorless (unless the specific peptide is normally colored). Lyophilized powders should appear as a uniform cake or powder without discoloration. Look for particulate matter, cloudiness, or any signs of vial damage.
- Check the shipping conditions. If the product requires refrigeration, was it shipped with adequate cold packing? Did the cold packs still have some remaining cold content when the package arrived? If the product appears to have been exposed to elevated temperatures during shipping, contact the pharmacy before using it.
- Request the COA. If the pharmacy doesn't automatically include a COA with your order, request one for your specific lot/batch number. Review it using the guidance provided earlier in this report.
- Store properly. Follow the storage instructions on the label. Most compounded peptide preparations require refrigeration (2-8 degrees Celsius). Some may be stored at room temperature for limited periods. Detailed storage guidance is available in the peptide storage and stability guide.
Proper Administration Technique
For injectable peptide preparations, proper administration technique is critical for safety:
- Hand hygiene: Wash your hands thoroughly before handling and administering any injectable medication
- Aseptic withdrawal: Swab the vial's rubber stopper with an alcohol prep pad before each withdrawal. Use a new, sterile syringe and needle for each dose
- Injection site preparation: Clean the injection site with an alcohol swab and allow it to dry before injecting
- Proper reconstitution: If your peptide is supplied as a lyophilized powder, follow the reconstitution instructions precisely. Use the correct diluent (typically bacteriostatic water or sterile water for injection), add it gently to avoid foaming, and swirl (don't shake) to dissolve. The reconstitution guide provides step-by-step instructions
- Dose accuracy: Use an insulin syringe with appropriate markings for accurate measurement of small volumes. Double-check the dose before injecting
Recognizing Adverse Reactions
While most peptide therapies are well tolerated, adverse reactions can occur. Some are expected pharmacological effects (such as nausea with GLP-1 agonists), while others may indicate a quality problem with the compounded product.
Signs that may indicate a product quality problem (as opposed to normal side effects):
- Injection site infection - redness, swelling, warmth, pain, or drainage at the injection site that develops or worsens over 24-72 hours
- Fever or chills developing shortly after injection (could indicate endotoxin exposure or microbial contamination)
- Allergic-type reactions - hives, difficulty breathing, swelling - especially if they occur with a product you've previously tolerated without issue from a different batch
- Complete lack of expected therapeutic effect when you've previously responded to the same peptide from a different batch (could indicate subpotent product)
- Exaggerated therapeutic effects or unexpected side effects (could indicate superpotent product)
If you experience any of these symptoms, stop using the product immediately and contact your healthcare provider. Save the vial and any remaining product - it may need to be tested to determine whether a quality problem exists.
Reporting Adverse Events
Adverse events related to compounded medications should be reported to multiple entities:
- Your healthcare provider: First and foremost, seek medical attention if needed and inform your prescriber
- The compounding pharmacy: Report the event to the pharmacy so they can investigate and potentially quarantine the affected batch
- The FDA: You can report adverse events directly to the FDA through the MedWatch program (online at www.fda.gov/medwatch or by phone at 1-800-FDA-1088). This report goes into the FDA's adverse event database and helps identify safety signals
- Your state board of pharmacy: Filing a complaint with the state board can trigger an investigation of the pharmacy's practices
Adverse event reporting isn't just about your individual situation - it helps protect other patients. When multiple reports about the same pharmacy or product accumulate, regulatory agencies can take action to address quality problems before more patients are harmed.
Figure 8: Patient safety flowchart for compounded peptide therapy - from receipt to administration and monitoring
Working with Your Healthcare Provider
Compounded peptide therapy should always be supervised by a qualified healthcare provider. Your provider plays several essential safety roles:
- Prescribing authority: Only a licensed prescriber can write a prescription for a compounded peptide. This requirement ensures that someone with medical training has evaluated whether the therapy is appropriate for your specific health situation
- Dose management: Your provider should establish the correct starting dose, titration schedule, and maintenance dose based on your individual response and health parameters
- Monitoring: Regular follow-up appointments and appropriate laboratory monitoring help ensure that the therapy is producing the desired effects without adverse consequences
- Pharmacy selection: Your provider may recommend or require specific compounding pharmacies based on their own evaluation of pharmacy quality. If your provider has established a relationship with a particular pharmacy, it likely means they've vetted that pharmacy's quality practices
- Adverse event management: If you experience adverse effects, your provider can determine whether they're expected pharmacological effects or potential indicators of product quality problems, and can adjust your treatment accordingly
Be wary of telehealth platforms that prescribe compounded peptides with minimal evaluation - a brief online questionnaire doesn't substitute for a thorough medical assessment. While telehealth can be an appropriate channel for prescribing when done properly (with a genuine provider-patient relationship, thorough medical history review, and appropriate follow-up), some platforms prioritize volume over patient safety. A comprehensive assessment should precede any peptide therapy prescription.
Choosing Your Pharmacy: A Practical Decision Framework
With all the information covered in this report, here's a practical framework for evaluating and selecting a compounding pharmacy for peptide therapy. This framework organizes the quality indicators into a priority hierarchy that helps you make a systematic decision.
Tier 1: Non-Negotiable Requirements
These are baseline requirements that any compounding pharmacy must meet before you should consider using them for injectable peptides:
- Valid state pharmacy license in good standing (no unresolved disciplinary actions)
- Requires a valid prescription from a licensed healthcare provider
- Performs sterility testing on injectable preparations
- Performs endotoxin testing on injectable preparations
- Compounds in a USP 797-compliant environment
- Willing to provide Certificates of Analysis upon request
If a pharmacy doesn't meet all of these requirements, do not use them for injectable peptide therapy, regardless of price, convenience, or marketing claims.
Tier 2: Strong Quality Indicators
These factors significantly increase confidence in product quality and represent best practices in the industry:
- 503B outsourcing facility registered with the FDA
- PCAB accreditation for sterile compounding
- Potency testing on every batch
- Third-party testing by an ISO 17025-accredited laboratory
- Stability data supporting assigned beyond-use dates
- FDA-registered API suppliers with full documentation
- Quarantine-until-release protocol (products held until all testing confirmed)
- Clean FDA inspection history (for 503B facilities)
A pharmacy that meets all Tier 1 requirements and most or all Tier 2 indicators represents a strong choice for compounded peptide therapy.
Tier 3: Additional Quality Differentiators
These factors further distinguish premium compounding operations:
- Specialized peptide compounding expertise and dedicated peptide compounding staff
- Multiple quality accreditations (PCAB + NABP, for example)
- Published or available stability data for specific peptide formulations
- Temperature-monitored cold chain shipping with data loggers
- Proactive COA distribution (COAs provided automatically with orders)
- Pharmacist consultation available for patient questions
- Voluntary adverse event reporting (for 503A pharmacies)
- Environmental monitoring trending data available upon request
Practical Evaluation Steps
- Start with verification. Before anything else, verify the pharmacy's license, registration, and accreditation status through official databases (state board, FDA, ACHC/PCAB).
- Ask the key questions. Use the questions from the earlier section to assess the pharmacy's quality practices. Pay attention not just to the answers but to how they're delivered - confidence and transparency are positive signs.
- Request a COA. Ask for a sample COA for a product similar to what you'll be ordering. Review it using the COA evaluation criteria discussed earlier.
- Compare thoughtfully. If you're considering multiple pharmacies, compare them on quality indicators first and price second. A pharmacy that's $50/month cheaper but can't provide COAs or doesn't perform endotoxin testing isn't actually a better value.
- Consult your provider. Share your research with your healthcare provider and ask for their input. They may have experience with specific pharmacies or additional quality concerns based on their clinical experience.
- Monitor ongoing quality. Once you've selected a pharmacy, continue to evaluate quality with each order. Check labels, inspect products, request periodic COAs, and report any concerns promptly.
THE BOTTOM LINE
Choosing a compounding pharmacy for peptide therapy is a consequential decision that deserves serious attention. The difference between a high-quality compounding pharmacy and a substandard one can affect the safety and efficacy of your treatment. Use the framework in this guide to evaluate your options systematically, verify claims independently, and select a pharmacy whose quality practices align with the standards that injectable medications demand. Your health is worth the extra effort. Visit the Peptide Hub for additional resources on peptide therapy, or take the free assessment to get started with a quality-focused pharmacy.
Frequently Asked Questions
References
- Allen LV Jr. The Art, Science, and Technology of Pharmaceutical Compounding. 5th ed. Washington, DC: American Pharmacists Association; 2016.
- Centers for Disease Control and Prevention (CDC). Multistate Outbreak of Fungal Meningitis and Other Infections. CDC HAI Outbreak Archives. 2015. Available at: https://archive.cdc.gov/www_cdc_gov/hai/outbreaks/meningitis.html
- Drug Quality and Security Act (DQSA). Public Law 113-54. 113th Congress. November 27, 2013.
- FDA. Compounding and the FDA: Questions and Answers. U.S. Food and Drug Administration. 2024. Available at: https://www.fda.gov/drugs/human-drug-compounding/compounding-and-fda-questions-and-answers
- FDA. FDA's Concerns with Unapproved GLP-1 Drugs Used for Weight Loss. U.S. Food and Drug Administration. 2025. Available at: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/fdas-concerns-unapproved-glp-1-drugs-used-weight-loss
- FDA. Human Drug Compounding Outsourcing Facility Registration and Drug Listing. U.S. Food and Drug Administration. 2024.
- FDA. Drug Products or Categories of Drug Products that Present Demonstrable Difficulties for Compounding Under Sections 503A or 503B. Proposed Rule. Federal Register. March 20, 2024.
- Gudeman J, Jozwiakowski M, Chollet J, Randell M. Potential Risks of Pharmacy Compounding. Drugs R D. 2013;13(1):1-8. doi:10.1007/s40268-013-0005-9
- Ghazal HS, Dyas AM, Ford JL, Hutcheon GA. Content Analysis of US FDA Warning Letters Issued to Compounding Pharmacies Regarding Violations of Current Good Manufacturing Practices Between 2017 and 2022. J Pharm Policy Pract. 2022;15:95. doi:10.1186/s40545-022-00493-7
- McElhiney LF. Compounding Sterile Preparations. 4th ed. Bethesda, MD: ASHP; 2020.
- Multistate Outbreak Investigation Team. Multistate fungal meningitis outbreak - preliminary report. N Engl J Med. 2013;369(22):2090-2098. doi:10.1056/NEJMoa1312629
- Pharmacy Compounding Accreditation Board (PCAB). Accreditation Standards for Compounding Pharmacies. ACHC. 2024. Available at: https://achc.org/pcab/
- Smith RM, Schaefer MK, Kainer MA, et al. Fungal infections associated with contaminated methylprednisolone injections. N Engl J Med. 2013;369(17):1598-1609. doi:10.1056/NEJMoa1213978
- Stephenson RC, Clarke S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J Biol Chem. 1989;264(11):6164-6170. doi:10.1016/S0021-9258(18)83328-6
- U.S. Department of Justice. Owner of New England Compounding Center Convicted of Racketeering Leading to Nationwide Fungal Meningitis Outbreak. Press Release. March 22, 2017.
- U.S. Pharmacopeia (USP). General Chapter <71> Sterility Tests. USP-NF. 2023.
- U.S. Pharmacopeia (USP). General Chapter <85> Bacterial Endotoxins Test. USP-NF. 2023.
- U.S. Pharmacopeia (USP). General Chapter <795> Pharmaceutical Compounding - Nonsterile Preparations. USP-NF. Revised November 1, 2023.
- U.S. Pharmacopeia (USP). General Chapter <797> Pharmaceutical Compounding - Sterile Preparations. USP-NF. Revised November 1, 2023.
- U.S. Pharmacopeia (USP). General Chapter <800> Hazardous Drugs - Handling in Healthcare Settings. USP-NF. 2020.
- Zapadka KL, Becher FJ, Gomes Dos Santos AL, Jackson SE. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus. 2017;7(6):20170030. doi:10.1098/rsfs.2017.0030
- Bak A, Leung D, Barrett SE, et al. Physicochemical and Formulation Developability Assessment for Therapeutic Peptide Delivery - A Primer. AAPS J. 2015;17(1):144-155. doi:10.1208/s12248-014-9688-2
- Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20(1):122-128. doi:10.1016/j.drudis.2014.10.003
- Lau JL, Dunn MK. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg Med Chem. 2018;26(10):2700-2707. doi:10.1016/j.bmc.2017.06.052
- National Association of Boards of Pharmacy (NABP). Compounding Pharmacy Accreditation Standards. NABP. 2024. Available at: https://nabp.pharmacy/programs/accreditations/compounding-pharmacy/
- Sellers S, Utian WH. Pharmacy compounding primer for physicians: prescriber beware. Drugs. 2012;72(16):2043-2050. doi:10.2165/11636830-000000000-00000
- Breckenridge A, Jacob R. Overcoming the legal and regulatory barriers to drug repurposing. Nat Rev Drug Discov. 2019;18(1):1-2. doi:10.1038/nrd.2018.92
- Thompson CA. FDA increases compounding pharmacy inspections. Am J Health Syst Pharm. 2014;71(5):362-363. doi:10.2146/news140017
- Caon T, Jin L, Simoes CM, Norton RS, Nicolazzo JA. Enhancing the Buccal Mucosal Delivery of Peptide and Protein Therapeutics. Pharm Res. 2015;32(1):1-21. doi:10.1007/s11095-014-1485-1
- FDA. FDA Issues Warning Letters to Compounders and Manufacturers of GLP-1 Products. FDA News Release. September 9, 2025.
- Patel A, Cholkar K, Mitra AK. Recent developments in protein and peptide parenteral delivery approaches. Ther Deliv. 2014;5(3):337-365. doi:10.4155/tde.14.5
- Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of protein pharmaceuticals: an update. Pharm Res. 2010;27(4):544-575. doi:10.1007/s11095-009-0045-6
- Lee VH. Enzymatic barriers to peptide and protein absorption. Crit Rev Ther Drug Carrier Syst. 1988;5(2):69-97. PMID: 3068521
- Michigan Attorney General. NECC Owner Barry Cadden Pleads to 11 Counts of Manslaughter. Press Release. March 5, 2024.
- Banga AK. Therapeutic Peptides and Proteins: Formulation, Processing, and Delivery Systems. 3rd ed. Boca Raton, FL: CRC Press; 2015. doi:10.1201/b18392
- Muheem A, Shakeel F, Jahangir MA, et al. A review on the strategies for oral delivery of proteins and peptides and their clinical perspectives. Saudi Pharm J. 2016;24(4):413-428. doi:10.1016/j.jsps.2014.06.004
- Anderson JR, Nawarskas JJ. Pharmacist-managed therapeutic drug monitoring of compounded medications. J Pharm Pract. 2016;29(4):411-415. doi:10.1177/0897190014566314