Executive Summary
The story of peptide medicine spans more than a century, from a desperate experiment in a Toronto laboratory in 1921 to a global weight-loss phenomenon in 2024. Along the way, peptides have saved hundreds of millions of lives, earned a dozen Nobel Prizes, and reshaped how we think about treating chronic disease.
Key Takeaways
- Insulin (1921) was the first peptide isolated for therapeutic use, and it remains one of the most prescribed drugs worldwide.
- Solid-phase peptide synthesis (1963) and recombinant DNA technology (1970s-80s) were the two enabling breakthroughs that made modern peptide drugs possible.
- GLP-1 receptor agonists, discovered in the 1980s and first approved in 2005, now represent the fastest-growing segment of the pharmaceutical market.
- More than 80 peptide drugs have received FDA approval, with the pace of approvals accelerating in the 2020s.
- The peptide therapeutics market is projected to grow from $38 billion (2023) to over $106 billion by 2033.
Peptides are relatively short chains of amino acids, typically defined as sequences containing fewer than approximately 50 residues in total length. They serve as hormones, neurotransmitters, growth factors, and antimicrobial agents throughout the human body. When Frederick Banting and Charles Best isolated insulin from canine pancreatic extracts in the summer of 1921, they didn't just discover a treatment for diabetes. They launched an entire field of medicine built on the idea that the body's own signaling molecules could be harnessed as drugs.
That idea has proven extraordinarily durable. Over the past 100 years, researchers have identified thousands of bioactive peptides, synthesized many of them in the laboratory, and turned dozens into approved therapeutics. The pace of discovery has accelerated with each passing decade. In the 1950s, Vincent du Vigneaud's synthesis of oxytocin demonstrated that a peptide hormone could be recreated from scratch. In the 1960s, Bruce Merrifield's solid-phase peptide synthesis (SPPS) method made it possible to build peptides quickly and reliably. In the 1980s, recombinant DNA technology allowed mass production of human insulin and growth hormone without relying on animal or cadaver sources.
Then came the incretin era. The discovery of glucagon-like peptide-1 (GLP-1) in the mid-1980s set the stage for what would become the most commercially successful class of peptide drugs in history. From the first approval of exenatide (Byetta) in 2005 to the explosive growth of semaglutide (Ozempic, Wegovy) and tirzepatide (Mounjaro, Zepbound) in the 2020s, GLP-1 receptor agonists have transformed the treatment of type 2 diabetes and obesity. The peptide therapeutics market reached approximately $38 billion in 2023, and projections suggest it could exceed $106 billion by 2033.
This report traces the complete arc of peptide medicine, from its origins in early 20th-century endocrinology through the chemistry breakthroughs of the mid-century, the biotechnology era, and the modern GLP-1 explosion. We'll examine the key figures, the critical experiments, the Nobel Prizes, and the regulatory milestones that have shaped this field. Whether you're a clinician, a researcher, or someone curious about how a class of molecules went from laboratory curiosity to front-page news, this history offers essential context for understanding where peptide medicine has been and where it's headed.
Key Takeaways
- Insulin (1921) was the first peptide isolated for therapeutic use, and it remains one of the most prescribed drugs worldwide.
- Solid-phase peptide synthesis (1963) and recombinant DNA technology (1970s-80s) were the two enabling breakthroughs that made modern peptide drugs possible.
- GLP-1 receptor agonists, discovered in the 1980s and first approved in 2005, now represent the fastest-growing segment of the pharmaceutical market.
- More than 80 peptide drugs have received FDA approval, with the pace of approvals accelerating in the 2020s.
- The peptide therapeutics market is projected to grow from $38 billion (2023) to over $106 billion by 2033.

1921-1950: The Insulin Era
Before insulin, a diagnosis of type 1 diabetes was a death sentence. Children wasted away on starvation diets, the only treatment available. The isolation of insulin in 1921 changed everything, and it established the template for peptide drug development that would be followed for the next hundred years.
The Problem: Diabetes Before Insulin
By the early 20th century, physicians understood that diabetes mellitus involved the pancreas. In 1889, German researchers Oskar Minkowski and Joseph von Mering had demonstrated that removing a dog's pancreas caused the animal to develop severe diabetes. Paul Langerhans had described clusters of specialized cells within the pancreas (later named the islets of Langerhans) as early as 1869. Edward Albert Sharpey-Schafer coined the name "insulin" in 1916 for the hypothetical substance produced by these islets that controlled blood sugar.
But nobody had managed to extract this substance in a form that could be administered to patients. Several researchers had tried and failed. Georg Zuelzer in Berlin, Ernest Scott in Chicago, Nicolae Paulescu in Bucharest - all came tantalizingly close. The pancreatic digestive enzymes tended to destroy the insulin during extraction, and purification techniques were primitive by modern standards.
Banting and Best: The Toronto Experiments
Frederick Banting was a young orthopedic surgeon in London, Ontario, with no research experience and a struggling practice. In October 1920, while preparing a lecture on the pancreas, he had an idea: if he ligated (tied off) the pancreatic ducts of dogs, the exocrine tissue producing digestive enzymes would atrophy, while the islets of Langerhans might survive. He could then extract the internal secretion without it being destroyed by trypsin and other proteases.
Banting brought his idea to J.J.R. Macleod, a professor of physiology at the University of Toronto and a recognized authority on carbohydrate metabolism. Macleod was skeptical but gave Banting laboratory space, ten dogs, and an assistant: Charles Best, a 22-year-old physiology and biochemistry student who won a coin toss with another student for the summer position.
On May 17, 1921, Banting and Best began their experiments. They ligated the pancreatic ducts of dogs, waited for the exocrine tissue to degenerate, and then prepared extracts from the remaining tissue. On July 27, 1921, they administered their extract (which they called "isletin") to a depancreatized diabetic dog. The dog's blood sugar dropped from 200 mg/dL to 120 mg/dL within two hours. They repeated the experiment multiple times with consistent results.
By the fall of 1921, Banting and Best had switched to using fetal calf pancreases (which have relatively more islet tissue and less exocrine tissue) and then whole beef pancreas. James Bertram Collip, a biochemist visiting from the University of Alberta, joined the team and developed a purification method using varying concentrations of alcohol to precipitate the active extract while removing toxic contaminants.
The First Human Treatment
On January 11, 1922, Leonard Thompson, a 14-year-old boy dying of diabetes at Toronto General Hospital, became the first person to receive an injection of pancreatic extract for the treatment of diabetes. The initial results were modest: his blood sugar dropped somewhat, but he developed a sterile abscess at the injection site. The extract was too impure. Collip went back to work, and on January 23, Thompson received a more refined preparation. This time, his blood sugar dropped from 520 mg/dL to 120 mg/dL. His ketoacidosis cleared. He gained weight and energy. He would live another 13 years.
The effect was so dramatic that clinicians began administering the extract to other patients almost immediately. By February 1922, six more patients were being treated at Toronto General Hospital. Word spread rapidly through the medical community. Banting and Best published their results in the Canadian Medical Association Journal in March 1922.
Scaling Up: Eli Lilly and Mass Production
The Toronto team quickly realized they couldn't produce enough insulin for all the patients who needed it. In May 1922, they entered into a collaboration with Eli Lilly and Company in Indianapolis. George B. Walden, Lilly's head chemist, discovered isoelectric precipitation, a technique that exploited the fact that insulin is least soluble at a specific pH (its isoelectric point). This allowed large-scale purification of insulin from animal pancreases with much higher yields and greater purity than the Toronto methods.
By the fall of 1922, Lilly was producing insulin in commercial quantities. In 1923, Novo Nordisk (then called Nordisk Insulinlaboratorium) began production in Denmark, and other manufacturers followed. Insulin was distributed across North America and Europe, saving thousands of lives within the first year of availability.
The Nobel Prize Controversy
In October 1923, the Nobel Prize in Physiology or Medicine was awarded to Banting and Macleod. The decision ignited a bitter controversy. Banting was furious that Best had been excluded, and he shared his prize money with Best. Macleod, in turn, shared his portion with Collip. The debate over credit continues to this day: Nicolae Paulescu in Romania had published results with pancreatic extracts before Banting, and some historians argue his contributions have been unfairly overshadowed.
Regardless of the credit disputes, the practical impact was clear. Insulin was the first peptide hormone to be isolated, purified, and used therapeutically. It established a model that would be repeated again and again: identify a biological signaling molecule, figure out how to produce it at scale, and administer it to patients whose bodies can't make enough of it on their own.
Insulin's Evolution: 1920s Through 1950s
The insulin available in the early 1920s was short-acting, requiring multiple daily injections. Patients and physicians quickly began looking for longer-acting formulations. In 1936, Hans Christian Hagedorn in Denmark developed protamine insulin, which combined insulin with protamine (a protein from fish sperm) to slow absorption. This became known as NPH insulin (Neutral Protamine Hagedorn) and remained a standard treatment for decades.
In 1951, the Grubb-Novo collaboration introduced Lente insulin, which used zinc rather than protamine to create intermediate and long-acting formulations. These developments gave physicians more flexibility in managing patients' blood sugar levels throughout the day and night.
Meanwhile, researchers were beginning to understand insulin's molecular structure. Dorothy Hodgkin used X-ray crystallography to determine insulin's three-dimensional structure in the 1930s and 1940s, work for which she would receive the Nobel Prize in Chemistry in 1964. Frederick Sanger began his landmark work on insulin's amino acid sequence in the mid-1940s, ultimately completing it in 1955. This was the first time the complete amino acid sequence of any protein had been determined, and it earned Sanger the Nobel Prize in Chemistry in 1958.
The Spread of Insulin Worldwide
The dissemination of insulin from Toronto to the rest of the world happened with remarkable speed for the era. By late 1922, physicians across North America were clamoring for access to the extract. The University of Toronto established a committee to oversee insulin distribution and quality control, recognizing that unregulated production could be dangerous. Banting, Best, and Collip assigned their patent rights to the University of Toronto for $1 each, explicitly choosing not to profit from the discovery.
In Europe, August Krogh, a Danish Nobel laureate who had visited Toronto to learn about insulin (his wife had diabetes), brought the technology back to Denmark. He helped establish the Nordisk Insulinlaboratorium in 1923, which would later become Novo Nordisk, the company that today produces semaglutide. The British Medical Research Council also obtained manufacturing rights and facilitated insulin production in the United Kingdom.
The global spread of insulin production revealed an important dynamic that would recur throughout peptide medicine history: the tension between rapid dissemination of life-saving treatments and the need for quality control. Early insulin preparations varied dramatically in potency. Some patients experienced severe hypoglycemia from overly concentrated batches, while others got little benefit from weak preparations. The development of standardized units (originally defined by biological assay in rabbits) and international reference standards helped address this problem, but it took several years before insulin quality became consistently reliable.
The Starvation Diet and Early Diabetes Management
Before insulin, the primary treatment for diabetes was dietary restriction. Frederick Allen, an American physician, was the leading proponent of what became known as the "starvation diet" or "Allen diet." Patients were restricted to as few as 400-500 calories per day in an attempt to reduce blood sugar levels. While this approach could extend life by months or even a year or two, it was grueling. Patients became skeletal, weak, and prone to infections. Many died of starvation rather than diabetes itself.
Elliott Joslin, who founded the Joslin Diabetes Center in Boston, was another prominent advocate of dietary management. When insulin became available, Joslin became one of its earliest and most enthusiastic adopters. He recognized that insulin didn't cure diabetes but transformed it into a disease that required ongoing management: regular injections, blood sugar monitoring, dietary attention, and exercise.
This concept of chronic disease management, which seems obvious today, was relatively novel in the 1920s. Most drugs of the era cured infections or relieved acute symptoms. Insulin was one of the first drugs that patients needed to take for the rest of their lives. It established the model for chronic disease therapeutics that would later apply to hypertension medications, statins, HIV antiretrovirals, and, of course, modern GLP-1 agonists.
The early decades of insulin therapy also revealed an important challenge that would recur throughout peptide medicine: the difficulty of precisely matching drug delivery to physiological need. The pancreas secretes insulin in a finely tuned pattern, with low basal secretion between meals and rapid bursts in response to food. Injected insulin, by contrast, is absorbed slowly and unpredictably from subcutaneous tissue, creating a mismatch between insulin levels and glucose demands. This mismatch leads to episodes of both hyperglycemia (too little insulin relative to glucose) and hypoglycemia (too much insulin relative to glucose), both of which have serious health consequences. The quest to improve insulin delivery, from longer-acting formulations to insulin pumps to closed-loop "artificial pancreas" systems, has been a central theme of diabetes technology development for a century. The same challenge applies to modern peptide drugs: matching the kinetics of drug delivery to the body's physiological requirements remains an active area of innovation.
Insulin and the Dawn of Clinical Biochemistry
The insulin era also catalyzed the development of clinical biochemistry as a discipline. Accurate measurement of blood glucose levels was essential for dosing insulin and monitoring treatment. Before insulin, blood glucose measurement was an obscure research technique. After insulin, it became a routine clinical procedure, and the development of rapid, reliable blood glucose assays drove innovation in laboratory medicine.
Herbert Hagedorn in Denmark developed the protamine zinc insulin (PZI) formulation by combining insulin with protamine and zinc, creating a longer-acting preparation that could reduce the number of daily injections. His work required careful titration and blood glucose monitoring, pushing the development of more practical testing methods. The concept of matching insulin action to the patient's glucose patterns (what we now call "intensive insulin therapy") emerged gradually through the 1930s and 1940s.
The development of self-monitoring blood glucose (SMBG) technologies in the 1970s and 1980s, and later continuous glucose monitoring (CGM) systems in the 2000s and 2010s, are direct descendants of the clinical imperative created by insulin therapy. Insulin pumps, first developed in the 1960s and refined into portable devices in the 1970s and 1980s, represent the other side of the technology equation: increasingly sophisticated systems for delivering the peptide drug.
Clinical Impact
Before insulin, children diagnosed with type 1 diabetes rarely survived more than a year or two. With insulin treatment, life expectancy increased dramatically. Leonard Thompson, the first patient treated, lived to age 27. Today, people with type 1 diabetes who manage their condition well can expect near-normal lifespans. The transformation from a fatal diagnosis to a manageable chronic condition remains one of medicine's greatest achievements.
| Year | Milestone | Key Figures |
|---|---|---|
| 1889 | Pancreatectomy shown to cause diabetes in dogs | Minkowski, von Mering |
| 1916 | Name "insulin" coined for hypothetical pancreatic hormone | Sharpey-Schafer |
| 1921 | Insulin isolated from canine pancreatic extracts | Banting, Best |
| 1922 | First human patient treated with insulin (Leonard Thompson) | Banting, Best, Collip, Macleod |
| 1922 | Isoelectric precipitation enables mass production | George Walden (Eli Lilly) |
| 1923 | Nobel Prize in Physiology or Medicine for insulin discovery | Banting, Macleod |
| 1936 | Protamine insulin (NPH) for longer action | Hagedorn |
| 1955 | Complete amino acid sequence of insulin determined | Frederick Sanger |
| 1958 | Nobel Prize in Chemistry for protein sequencing | Sanger |
1950-1980: Peptide Hormone Discovery
The three decades following World War II saw an explosion of peptide hormone discoveries. Researchers identified, sequenced, and synthesized dozens of biologically active peptides, while chemists developed the tools needed to build these molecules from scratch. This era produced at least five Nobel Prizes directly related to peptide science.
Vincent du Vigneaud and the Synthesis of Oxytocin (1953)
The first peptide hormone to be chemically synthesized was oxytocin. Vincent du Vigneaud, a biochemist at Cornell University Medical College, had spent decades studying sulfur-containing biological compounds, including the amino acid cystine and the vitamin biotin. His interest in sulfur chemistry led him naturally to oxytocin and vasopressin, two peptide hormones from the posterior pituitary gland that contain disulfide bonds.
Du Vigneaud's group first determined the complete amino acid sequence of oxytocin, a nine-amino-acid peptide with an internal disulfide bridge between two cysteine residues. In October 1953, he announced the successful total synthesis of oxytocin. The synthetic version was biologically indistinguishable from the natural hormone: it stimulated uterine contractions and promoted milk ejection in postpartum animals with the same potency as the extracted material.
This was a landmark achievement. For the first time, a peptide hormone had been recreated entirely from chemical building blocks. The synthesis confirmed that biological activity depended on the precise sequence and arrangement of amino acids, not on some mysterious "vital force" associated with living tissue. Du Vigneaud received the Nobel Prize in Chemistry in 1955 for this work.
Following oxytocin, du Vigneaud's laboratory also synthesized vasopressin (antidiuretic hormone), another nine-amino-acid peptide that differs from oxytocin at only two positions. Both synthetic hormones eventually found clinical applications: synthetic oxytocin (Pitocin) for labor induction and postpartum hemorrhage, and synthetic vasopressin (desmopressin) for diabetes insipidus and bedwetting.
The Peptide Hormone Boom: ACTH, Glucagon, and Beyond
Du Vigneaud's success with oxytocin inspired a wave of peptide hormone research. Through the 1950s and 1960s, researchers identified and characterized an ever-growing list of biologically active peptides:
- Adrenocorticotropic hormone (ACTH): A 39-amino-acid peptide from the anterior pituitary that stimulates cortisol production. Its sequence was determined in 1956 by C.H. Li and colleagues, and it was synthesized by Klaus Hofmann in 1960.
- Glucagon: A 29-amino-acid peptide produced by pancreatic alpha cells that raises blood sugar. Its sequence was established by Bromer and colleagues in 1957.
- Calcitonin: A 32-amino-acid peptide from the thyroid gland that regulates calcium metabolism. Discovered in 1962 by Harold Copp.
- Gastrin: A peptide hormone that stimulates stomach acid secretion. Identified by Rod Gregory and Hilda Tracy in 1964.
- Secretin: Originally discovered in 1902 by Bayliss and Starling (making it the first hormone ever identified), its complete sequence was finally determined in 1966.
- Substance P: An 11-amino-acid neuropeptide involved in pain signaling and inflammation. First detected in 1931 by Ulf von Euler and John Gaddum, its full sequence was established in 1971 by Susan Leeman and Michael Chang.
The pace of peptide hormone discovery during this period was extraordinary. New peptides were being identified at a rate of several per year, each one revealing previously unknown regulatory mechanisms in the body. The tools enabling this discovery wave included improved protein purification techniques (gel filtration, ion exchange chromatography, affinity chromatography), amino acid analysis methods (including the Edman degradation sequencer, commercialized in the 1960s), and the radioimmunoassay technique developed by Yalow and Berson.
The diversity of biological functions served by peptide hormones was surprising. Peptides regulated blood pressure (angiotensin), water balance (vasopressin), milk production (oxytocin), growth (growth hormone, GHRH), stress responses (ACTH, CRH), reproduction (GnRH), calcium metabolism (calcitonin, PTH), digestion (gastrin, secretin, CCK), pain (enkephalins, endorphins), and appetite (cholecystokinin, neuropeptide Y). No other class of signaling molecules displayed such extraordinary functional diversity.
Each of these discoveries expanded the understanding of how peptide signaling molecules regulate physiology. The concept of "peptide hormones" broadened into "regulatory peptides" as researchers realized these molecules weren't limited to the classical endocrine glands. The gut, the brain, the immune system, the skin - virtually every tissue in the body produced peptide signals.
Bruce Merrifield and Solid-Phase Peptide Synthesis (1963)
While biologists were discovering new peptides at a rapid clip, chemists faced a practical problem. Synthesizing peptides in solution was painfully slow. Each coupling step required purification of intermediates, and yields dropped multiplicatively with each additional amino acid. Building a 20-residue peptide might take months of painstaking work.
Robert Bruce Merrifield, a biochemist at The Rockefeller Institute (now Rockefeller University), conceived an elegant solution in 1959. Instead of synthesizing peptides in solution, he proposed anchoring the growing peptide chain to an insoluble polymer bead. After each coupling reaction, impurities and excess reagents could be washed away simply by filtering the resin. No intermediate purification was needed. The peptide would be cleaved from the resin only after the complete sequence had been assembled.
Merrifield published his method in the Journal of the American Chemical Society in 1963. The paper described the synthesis of a tetrapeptide, Leu-Ala-Gly-Val, using a chloromethylated polystyrene resin as the solid support. The approach worked beautifully: yields were high, the process was fast, and it could be automated.
Merrifield and his colleagues quickly demonstrated the power of SPPS by synthesizing increasingly complex peptides. In the mid-1960s, they produced bradykinin (a 9-amino-acid vasodilator), angiotensin (an 8-amino-acid blood pressure regulator), desamino-oxytocin, and insulin. In 1969, Merrifield and Bernd Gutte announced the first synthesis of an enzyme - ribonuclease A, a 124-amino-acid protein - using the solid-phase method. This was a remarkable technical achievement, proving that SPPS could handle even full-length proteins.
The impact on peptide research was immense. Before SPPS, making a single peptide could occupy a chemist for an entire career. After SPPS, a competent technician could synthesize a 30-residue peptide in a few days. The method opened the floodgates for structure-activity relationship studies, enabling researchers to create hundreds of analogs of a given peptide and test each one for biological activity.
Merrifield received the Nobel Prize in Chemistry in 1984 for his invention. His original 1963 paper remains one of the five most cited publications in the history of the Journal of the American Chemical Society. In 2006, the ACS Division of the History of Chemistry recognized it as a Chemical Breakthrough Publication, a designation reserved for papers that have had a transformative impact on the field.
The practical impact of SPPS extended well beyond academic research. It enabled the pharmaceutical industry to pursue peptide drug development at a scale that would have been unthinkable with solution-phase methods. Consider the development path for a typical peptide drug candidate: hundreds of analogs must be synthesized and tested during the structure-activity relationship (SAR) phase, followed by optimization of lead compounds for stability, selectivity, and pharmacokinetics, then scale-up for preclinical toxicology studies, and finally manufacturing for clinical trials and eventual commercialization. Without SPPS, this process would take decades for a single compound. With SPPS, it can be completed in 5-10 years.
Merrifield's contribution also had an important philosophical dimension. By demonstrating that complex biological molecules could be built reliably from simple chemical building blocks using a systematic, automated process, he bridged the gap between chemistry and biology in a new way. The "information" in a peptide, its amino acid sequence, could be translated from a written formula into a physical molecule with complete fidelity. This concept, that biological information can be precisely replicated by chemical synthesis, was essential for the biotechnology era that followed.
Why SPPS Mattered
Solid-phase peptide synthesis didn't just speed up peptide chemistry. It fundamentally changed the relationship between biologists and chemists. Once a new peptide hormone was sequenced, chemists could synthesize it within weeks rather than years. This made it practical to study structure-activity relationships, to create modified analogs with improved properties (longer half-life, greater selectivity, oral bioavailability), and ultimately to develop peptide drugs. Without SPPS, the modern peptide therapeutics industry would not exist.
Roger Guillemin, Andrew Schally, and the Hypothalamic Peptides
One of the most dramatic chapters in peptide science was the fierce rivalry between Roger Guillemin and Andrew Schally over the hypothalamic releasing hormones. Both men spent decades trying to isolate and characterize the tiny peptides produced by the hypothalamus that control the anterior pituitary gland.
The work was heroically difficult. The hypothalamus is small, and these peptides are present in vanishingly low concentrations. Guillemin's group at Baylor (and later the Salk Institute) processed over five million sheep hypothalami. Schally's group at Tulane and the Veterans Administration processed more than two million pig hypothalami. The quantities of material they worked with were staggering, and the personal and professional rivalry between the two men was intense.
In 1969, Schally's team reported the isolation and characterization of thyrotropin-releasing hormone (TRH), a tripeptide (pyroGlu-His-Pro) that stimulates the pituitary to release thyroid-stimulating hormone. Guillemin's group independently published the same structure shortly afterward. TRH was the first hypothalamic releasing hormone to be fully characterized.
In 1971, Schally's group published the structure of gonadotropin-releasing hormone (GnRH, also called LHRH), a decapeptide that controls the release of luteinizing hormone and follicle-stimulating hormone. GnRH would later become therapeutically important: synthetic GnRH analogs (leuprolide, goserelin, nafarelin) are widely used in the treatment of prostate cancer, endometriosis, precocious puberty, and infertility.
Guillemin's group subsequently isolated somatostatin (growth hormone-inhibiting hormone) in 1973, a 14-amino-acid cyclic peptide, and growth hormone-releasing hormone (GHRH) in 1982. Somatostatin analogs like octreotide later became important drugs for treating acromegaly, carcinoid tumors, and other conditions.
Guillemin and Schally shared the 1977 Nobel Prize in Physiology or Medicine (along with Rosalyn Yalow, who developed radioimmunoassay, a technique critical for measuring tiny amounts of peptide hormones in blood). The prize recognized work that had profound implications for both basic science and clinical medicine. The hypothalamic peptides and their analogs have generated billions of dollars in pharmaceutical revenue and continue to be used in clinical practice today.
The clinical impact of hypothalamic peptide discoveries has been immense. GnRH analogs alone represent a multi-billion dollar drug class. Leuprolide acetate (Lupron, approved in 1985), the most widely used GnRH agonist, is prescribed for prostate cancer, endometriosis, uterine fibroids, central precocious puberty, and as part of in vitro fertilization (IVF) protocols. GnRH antagonists like cetrorelix and ganirelix offer a faster onset of gonadotropin suppression and are widely used in assisted reproduction.
Somatostatin analogs have similarly broad applications. Octreotide (Sandostatin, 1988) and lanreotide (Somatuline, 2007) are used for acromegaly (growth hormone excess), neuroendocrine tumors, variceal bleeding, and certain diarrheal diseases. Pasireotide (Signifor, 2012), a somatostatin analog with broader receptor subtype binding, was approved for Cushing's disease. These drugs demonstrate the power of peptide analog design: by modifying the natural 14-amino-acid somatostatin sequence, chemists created compounds with vastly improved half-lives (from 3 minutes for somatostatin to 90 minutes for octreotide to days for depot formulations), while retaining the desired biological activity and minimizing unwanted effects.
GHRH analogs, based on Guillemin's 1982 discovery of growth hormone-releasing hormone, have been used diagnostically to test pituitary function and therapeutically to stimulate growth hormone secretion. Tesamorelin (Egrifta), a synthetic GHRH analog, was approved in 2010 for the treatment of HIV-associated lipodystrophy, a condition in which antiretroviral drugs cause abnormal fat accumulation in the abdomen. The approval illustrated how a peptide hormone discovered through basic neuroendocrine research could eventually find a clinical application in an entirely different disease context.
Rosalyn Yalow and Radioimmunoassay
Rosalyn Yalow's contribution deserves special attention. Working with Solomon Berson at the Bronx VA Hospital, Yalow developed the radioimmunoassay (RIA) technique in the late 1950s. RIA exploits the specificity of antibodies: by combining a known amount of radiolabeled peptide with a specific antibody and then adding the sample to be measured, researchers could determine peptide concentrations in blood with extraordinary sensitivity, often detecting picomolar quantities.
Yalow and Berson first applied RIA to measure insulin levels in diabetic patients, demonstrating that type 2 diabetes was characterized by insulin resistance (adequate or elevated insulin levels) rather than insulin deficiency. This was a fundamental insight that reshaped the understanding of diabetes.
RIA was then adapted to measure virtually every known peptide hormone, from ACTH to GnRH to gastrin to somatostatin. It became an indispensable tool for endocrinology and was critical for the hypothalamic hormone work of Guillemin and Schally. Without the ability to measure trace amounts of peptides accurately, their decades of extraction and purification work would have been impossible.
Yalow was the second woman to win the Nobel Prize in Physiology or Medicine. Berson had died in 1972 and was ineligible for the prize, but Yalow consistently credited him as an equal partner in the work.
The Endorphins and Enkephalins: Peptides of Pain and Pleasure
In 1975, John Hughes and Hans Kosterlitz at the University of Aberdeen discovered two small peptides in pig brain extracts that bound to opioid receptors: Met-enkephalin and Leu-enkephalin, each only five amino acids long. These were the first "endogenous opioids" to be identified, and they launched a wave of research into the body's pain-modulation systems.
Shortly afterward, several groups (including those of C.H. Li, Roger Guillemin, and Avram Goldstein) identified larger endogenous opioid peptides: beta-endorphin (31 amino acids), dynorphin, and others. These peptides bound to the same receptors targeted by morphine and other opioid drugs, explaining how the body naturally modulates pain, mood, and reward.
Natriuretic Peptides and the Heart-Peptide Connection
In 1981, Adolfo de Bold at Queen's University in Canada made a surprising discovery: extracts from atrial heart tissue caused a dramatic increase in sodium excretion (natriuresis) and urine output when injected into rats. He had found atrial natriuretic peptide (ANP), a 28-amino-acid peptide hormone produced by heart atrial cells in response to stretching caused by increased blood volume.
ANP was the first peptide hormone found to be produced by the heart, overturning the assumption that the heart was purely a mechanical pump. It acts on the kidneys to promote sodium and water excretion, on blood vessels to cause vasodilation, and on the adrenal glands to suppress aldosterone secretion. The net effect is a reduction in blood pressure and blood volume, a natural counter-regulatory mechanism that helps prevent fluid overload.
Subsequently, brain natriuretic peptide (BNP, actually produced mainly by the ventricles despite its name) and C-type natriuretic peptide (CNP) were identified. BNP and its N-terminal fragment (NT-proBNP) became crucial diagnostic biomarkers for heart failure: elevated blood levels indicate that the heart is under stress, and serial measurements help guide treatment decisions.
Nesiritide (Natrecor), a recombinant form of BNP, was approved by the FDA in 2001 for acute decompensated heart failure. While its clinical use has been debated (the ASCEND-HF trial showed it relieved dyspnea but didn't improve survival), the natriuretic peptide system remains an active area of cardiovascular research. Vosoritide, the C-type natriuretic peptide analog approved for achondroplasia, demonstrates the therapeutic versatility of the natriuretic peptide family beyond its cardiovascular origins.
Peptide Antibiotics: Vancomycin, Daptomycin, and Beyond
Not all therapeutically important peptides are hormones. Peptide antibiotics have played a critical role in treating bacterial infections, particularly those caused by resistant organisms. Vancomycin, a glycopeptide antibiotic isolated from Amycolatopsis orientalis in 1956, has been a cornerstone treatment for methicillin-resistant Staphylococcus aureus (MRSA) and other serious Gram-positive infections for decades. Its mechanism involves binding to the D-Ala-D-Ala terminus of cell wall peptidoglycan precursors, preventing cell wall synthesis.
Daptomycin (Cubicin), a cyclic lipopeptide antibiotic approved in 2003, works by a completely different mechanism: it inserts into bacterial cell membranes in a calcium-dependent manner, forming pores that depolarize the membrane and kill the cell. This membrane-targeting mechanism means that bacteria have difficulty developing resistance, since the fundamental structure of their membranes can't be easily altered without compromising viability.
Polymyxins (colistin, polymyxin B), cyclic peptide antibiotics discovered in the 1940s, have experienced a resurgence as "drugs of last resort" against multi-drug-resistant Gram-negative bacteria. Although their nephrotoxicity limits use, they remain essential when no other antibiotics are effective.
The threat of antimicrobial resistance has renewed interest in antimicrobial peptides (AMPs) as a new class of antibiotics. Hundreds of AMPs have been identified in nature, from human defensins and cathelicidins to frog skin peptides and insect hemolymph factors. These peptides typically kill bacteria by disrupting their cell membranes through multiple simultaneous mechanisms, making it much harder for bacteria to develop resistance. Several synthetic AMPs are in clinical development, and if successful, they could represent a new chapter in peptide therapeutics.
The discovery of endogenous opioid peptides had wide-ranging implications. It deepened the understanding of pain physiology, addiction, stress responses, and the placebo effect. It also raised the tantalizing possibility of developing peptide-based painkillers that might be effective without the addiction liability of traditional opioids. While that goal has proven elusive, research into opioid peptides continues to this day.
The opioid peptide story also illustrates an important theme in peptide biology: the concept of post-translational processing. Beta-endorphin, met-enkephalin, and other opioid peptides are not produced directly from their own genes. Instead, they are cleaved from larger precursor proteins (pro-opiomelanocortin for beta-endorphin, proenkephalin for the enkephalins, prodynorphin for dynorphin) by specific processing enzymes. The same precursor protein can produce different peptide products in different tissues, depending on which processing enzymes are expressed. This "one gene, many peptides" principle, first elucidated in the opioid peptide system, turned out to be a general feature of peptide biology. The proglucagon gene, for example, produces glucagon in the pancreas but GLP-1 and GLP-2 in the intestine, through the action of different processing enzymes in each tissue.
Angiotensin and the Renin-Angiotensin System
The renin-angiotensin system (RAS) provides another instructive example of peptide biology translating into therapeutics, though in this case the drugs that resulted were not peptides themselves but rather inhibitors of peptide-generating enzymes. Angiotensin II, an 8-amino-acid peptide, is one of the most potent vasoconstrictors known. It's produced by a two-step enzymatic cascade: renin (an enzyme released by the kidneys) cleaves angiotensinogen (a liver protein) to produce angiotensin I (a 10-amino-acid peptide), and then angiotensin-converting enzyme (ACE, found primarily in the lungs) removes two amino acids from angiotensin I to produce angiotensin II.
Understanding this peptide cascade led to two of the most important drug classes in cardiovascular medicine: ACE inhibitors (captopril, enalapril, lisinopril, and many others) and angiotensin receptor blockers (ARBs, such as losartan and valsartan). ACE inhibitors, first developed in the 1970s based on peptides found in Brazilian pit viper venom (yet another example of natural peptides inspiring drug development), block the conversion of angiotensin I to angiotensin II. ARBs block the angiotensin II receptor directly. Both classes are now among the most widely prescribed cardiovascular drugs worldwide.
The captopril story deserves special mention. In 1965, Sergio Ferreira, a Brazilian pharmacologist, discovered that the venom of the pit viper Bothrops jararaca contained peptides that enhanced the hypotensive effect of bradykinin by inhibiting the enzyme that degrades it. This enzyme turned out to be the same as angiotensin-converting enzyme. John Vane's group at the Royal College of Surgeons in London, and independently David Cushman and Miguel Ondetti at Squibb (now Bristol-Myers Squibb), used this peptide lead to develop captopril, the first ACE inhibitor, approved in 1981. The story parallels the exendin-4/exenatide story: a peptide from animal venom points the way to a blockbuster drug class.
Neuropeptides and Brain Function
The 1970s and 1980s also saw an explosion of neuropeptide research. Substance P, vasopressin, oxytocin, neuropeptide Y, cholecystokinin (CCK), vasoactive intestinal peptide (VIP), calcitonin gene-related peptide (CGRP), galanin, and dozens of other peptide neurotransmitters and neuromodulators were identified and characterized. This work revealed that the brain uses peptides as signaling molecules far more extensively than had been appreciated.
Several of these neuropeptide discoveries have led to important drugs. CGRP antagonists (monoclonal antibodies like erenumab, fremanezumab, and galcanezumab, and small-molecule antagonists like rimegepant and ubrogepant) have become first-line treatments for migraine prevention and acute treatment. These drugs were developed based on decades of research showing that CGRP levels rise during migraine attacks and that CGRP receptor activation contributes to the pain and neuroinflammation of migraine.
Substance P and its receptor, the neurokinin 1 (NK1) receptor, are involved in pain signaling, inflammation, and the emetic (vomiting) reflex. Aprepitant (Emend), an NK1 receptor antagonist, was approved in 2003 for the prevention of chemotherapy-induced nausea and vomiting. It was the first clinically successful drug to target a neuropeptide receptor system.
Neuropeptide Y (NPY), one of the most abundant neuropeptides in the brain, is involved in appetite regulation, stress responses, anxiety, and circadian rhythms. Although NPY receptor-targeted drugs have not yet reached the market, the peptide's role in appetite control connects it to the broader incretin and GLP-1 story. The brain's appetite-regulating circuits involve complex interactions between GLP-1, NPY, melanocortins (alpha-MSH), agouti-related protein (AgRP), and other peptide signals. Understanding these interactions is essential for developing the next generation of anti-obesity therapeutics.
| Year | Discovery | Researchers | Nobel Prize |
|---|---|---|---|
| 1953 | First synthesis of a peptide hormone (oxytocin) | du Vigneaud | Chemistry, 1955 |
| 1955 | Insulin amino acid sequence determined | Sanger | Chemistry, 1958 |
| 1959 | Radioimmunoassay developed | Yalow, Berson | Physiology/Medicine, 1977 |
| 1963 | Solid-phase peptide synthesis (SPPS) | Merrifield | Chemistry, 1984 |
| 1964 | Insulin 3D structure (X-ray crystallography) | Hodgkin | Chemistry, 1964 |
| 1969 | TRH structure determined | Schally; Guillemin | Physiology/Medicine, 1977 |
| 1971 | GnRH structure determined | Schally | Physiology/Medicine, 1977 |
| 1973 | Somatostatin isolated | Guillemin | - |
| 1975 | Enkephalins discovered | Hughes, Kosterlitz | - |
1980-2000: Recombinant Technology
The development of recombinant DNA technology in the 1970s and 1980s transformed peptide medicine. For the first time, human peptide hormones could be produced in unlimited quantities using engineered bacteria or yeast cells, eliminating the need for animal or cadaver sources and dramatically reducing the risk of contamination.
Recombinant Human Insulin: The First Biotech Drug
For sixty years, patients with diabetes relied on insulin extracted from pig and cow pancreases. Animal insulin worked well for most patients, but it wasn't identical to human insulin. Porcine insulin differs from human insulin by one amino acid; bovine insulin by three. Some patients developed allergic reactions or antibodies to animal insulin, and the supply was fundamentally limited by the availability of animal pancreases from slaughterhouses.
In 1978, a team at the City of Hope National Medical Center, working with the biotechnology startup Genentech, used recombinant DNA technology to produce synthetic human insulin in E. coli bacteria. Arthur Riggs and Keiichi Itakura designed synthetic genes encoding the A and B chains of human insulin, which were separately expressed in bacteria and then combined to form the complete insulin molecule.
At almost the same time, Walter Gilbert's group at Harvard was pursuing an alternative approach, cloning the actual human insulin gene from human DNA. Both groups published their results in 1978-1979, setting off a race to commercialize recombinant human insulin.
Eli Lilly partnered with Genentech and received FDA approval for recombinant human insulin (Humulin) in October 1982. This was the first recombinant DNA drug product approved for human use. It was a watershed moment not just for diabetes treatment but for the entire pharmaceutical industry. Humulin demonstrated that genetically engineered organisms could safely and reliably produce human proteins at industrial scale.
The transition from animal to recombinant insulin took about a decade. By the early 1990s, most insulin-dependent patients in developed countries had switched to recombinant human insulin. The unlimited supply also opened the door to engineering insulin analogs with modified properties. Eli Lilly introduced lispro (Humalog) in 1996, the first rapid-acting insulin analog, which had its amino acid sequence slightly modified to prevent the self-association that slows absorption of regular insulin. Novo Nordisk followed with insulin aspart (NovoRapid/Novolog) in 2000 and insulin glargine (Lantus, by Sanofi) provided a true once-daily basal insulin in 2000.
Recombinant Human Growth Hormone: Eliminating a Deadly Risk
The story of recombinant growth hormone is even more dramatic. From the 1960s through the mid-1980s, children with growth hormone deficiency were treated with growth hormone extracted from the pituitary glands of human cadavers. The National Hormone and Pituitary Program (NHPP) in the United States coordinated the collection and distribution of cadaver-derived growth hormone, processing tens of thousands of pituitary glands per year.
In 1985, the program came to an abrupt halt. Four young adults who had received cadaver-derived growth hormone in the 1960s and 1970s developed Creutzfeldt-Jakob disease (CJD), a fatal prion disease. The pituitary glands used in the program had evidently been contaminated with prions from donors who had undiagnosed CJD. On April 19, 1985, distribution of pituitary-derived growth hormone was suspended in the United States. Ultimately, more than 200 cases of CJD were linked to cadaver-derived growth hormone worldwide.
The timing was fortunate in one respect: Genentech had been developing recombinant human growth hormone (somatropin) and had already begun clinical trials. On October 18, 1985, just six months after the pituitary GH recall, the FDA approved Genentech's Protropin (somatrem, a methionyl form of human growth hormone) for the treatment of growth hormone deficiency in children. Eli Lilly's Humatrope (true recombinant human growth hormone, somatropin) followed in 1987.
Recombinant growth hormone eliminated the risk of prion transmission and provided an unlimited supply. This allowed the indications to expand well beyond classic growth hormone deficiency. Growth hormone is now approved for Turner syndrome, chronic renal insufficiency, Prader-Willi syndrome, short stature associated with SHOX gene deficiency, idiopathic short stature, and adult growth hormone deficiency. The worldwide market for recombinant growth hormone products exceeds $4 billion annually.
The Impact of Recombinant Technology on Drug Safety
The transition from extracted to recombinant peptide drugs had safety implications that extended far beyond eliminating prion risk. Animal-derived insulins could trigger immune responses in some patients, leading to insulin allergy, insulin resistance due to antibody formation, and injection site reactions. Recombinant human insulin, being identical in sequence to the patient's own insulin, dramatically reduced these immunological complications.
Recombinant technology also eliminated the variability inherent in biological extraction. The potency of insulin extracted from animal pancreases could vary from batch to batch, depending on the source animals, the extraction conditions, and the purification methods. Recombinant insulin, produced from a single cloned gene under controlled fermentation conditions, was far more consistent in potency and purity. This consistency improved clinical outcomes by making dosing more predictable.
The environmental and ethical benefits were significant as well. Producing insulin from animal pancreases required millions of pig and cow pancreases annually, sourced from slaughterhouses. Recombinant production eliminated this dependency on the meat industry and removed a potential source of supply disruption (a disease outbreak affecting livestock, for example, could have interrupted the insulin supply). For patients with religious or ethical objections to animal-derived products, recombinant human insulin provided a welcome alternative.
The regulatory framework for recombinant drugs was established largely through the insulin and growth hormone approvals. The FDA developed guidelines for the production, characterization, and clinical testing of recombinant protein drugs that became the template for all subsequent biologic drug approvals. These guidelines addressed issues like cell bank characterization, process validation, product consistency testing, immunogenicity assessment, and post-market surveillance, all of which remain central to biologic drug regulation today.
Insulin Analog Design: Improving on Nature
Once recombinant technology made it possible to produce human insulin in unlimited quantities, researchers turned their attention to making insulin better. The natural insulin molecule, while effective, has several pharmacokinetic limitations. Regular human insulin forms hexamers (six-molecule aggregates) at pharmaceutical concentrations, and these hexamers must dissociate into monomers before the insulin can be absorbed from the injection site. This dissociation process takes 30-60 minutes, creating a delay between injection and onset of action that doesn't match the rapid glucose rise after a meal.
The first insulin analog designed to address this problem was insulin lispro (Humalog), developed by Eli Lilly and approved in 1996. Lispro has two amino acid positions reversed compared to human insulin (proline at position B28 and lysine at B29, instead of the natural lysine-proline sequence). This small change disrupts hexamer formation, allowing lispro to exist primarily as monomers and dimers at the injection site. The result is faster absorption, a quicker onset of action (15-30 minutes versus 30-60 minutes for regular insulin), and a shorter duration of action, all of which better match the post-meal glucose profile.
Insulin aspart (NovoRapid/NovoLog), approved in 2000, achieves similar rapid action by replacing proline at B28 with aspartic acid. Insulin glulisine (Apidra), approved in 2004, uses asparagine at B3 and lysine at B29 (replacing lysine at B3 and proline at B29). All three rapid-acting analogs produce faster onset and shorter duration than regular human insulin.
On the other end of the spectrum, long-acting insulin analogs were engineered for extended, flat pharmacokinetic profiles. Insulin glargine (Lantus), approved in 2000, has two extra arginine residues added to the B-chain C-terminus and asparagine at A21 replaced with glycine. These changes shift the isoelectric point to neutral pH, causing the analog to precipitate in the neutral pH of subcutaneous tissue and form a depot that slowly releases insulin over 24 hours. Insulin detemir (Levemir), approved in 2004, uses fatty acid acylation (a C14 myristic acid chain at B29) to enable albumin binding. Insulin degludec (Tresiba), approved in 2015, uses a C16 hexadecanedioic acid with a gamma-glutamic acid linker, forming multi-hexamer chains at the injection site that provide ultra-long-acting insulin release with a half-life of over 25 hours.
The engineering principles developed for insulin analogs, particularly the use of amino acid substitutions to alter self-association and fatty acid acylation for albumin binding, directly informed the later development of long-acting GLP-1 agonists. Novo Nordisk's expertise in lipidated insulin analogs (detemir, degludec) was directly translated to liraglutide and semaglutide. The same R&D team and the same pharmacological concepts were applied to a different peptide hormone with transformative results.
Other Recombinant Peptide Hormones
The success of recombinant insulin and growth hormone spurred development of other recombinant peptide and protein therapeutics throughout the 1980s and 1990s:
- Erythropoietin (EPO): A 165-amino-acid glycoprotein hormone that stimulates red blood cell production. Amgen's recombinant epoetin alfa (Epogen, Procrit) was approved in 1989 for anemia associated with chronic kidney disease. EPO became one of the best-selling biologic drugs, with peak sales exceeding $10 billion annually.
- Calcitonin: Synthetic salmon calcitonin (Miacalcin) was approved in 1984 for Paget's disease and later for osteoporosis. A nasal spray formulation followed in 1995.
- GnRH analogs: Leuprolide (Lupron, 1985), goserelin (Zoladex, 1989), and nafarelin (Synarel, 1990) were approved for prostate cancer, endometriosis, and precocious puberty. These are synthetic analogs of the natural GnRH decapeptide, modified for increased potency and duration of action.
- Octreotide: A synthetic octapeptide analog of somatostatin approved in 1988 for acromegaly and carcinoid tumors. Somatostatin itself has a half-life of only about 3 minutes; octreotide, through strategic amino acid substitutions, achieves a half-life of about 90 minutes.
- Desmopressin: A synthetic analog of vasopressin used for diabetes insipidus, bedwetting, and hemophilia A. Modified to enhance antidiuretic activity while reducing vasopressor effects.
The Emergence of Peptide Design Principles
The 1980s and 1990s also saw the emergence of rational peptide drug design. Researchers began to understand the principles that governed peptide stability, receptor binding, and pharmacokinetics, and they used this knowledge to engineer peptides with improved therapeutic properties.
Key strategies included:
- D-amino acid substitution: Replacing natural L-amino acids with their D-enantiomers at positions susceptible to protease cleavage. This made peptides resistant to enzymatic degradation without necessarily disrupting receptor binding.
- Cyclization: Creating circular peptides by forming bonds between the N-terminus and C-terminus, or between side chains. Cyclic peptides are generally more resistant to proteases and often have better membrane permeability.
- PEGylation: Attaching polyethylene glycol (PEG) chains to peptides to increase their molecular weight, reduce renal clearance, and extend half-life.
- Fatty acid acylation: Attaching lipid chains to peptides so they bind to serum albumin, which acts as a depot and dramatically extends circulating half-life. This approach would become crucial for the development of long-acting GLP-1 agonists.
- Unnatural amino acid incorporation: Using amino acid analogs not found in nature to improve metabolic stability, enhance receptor affinity, or add new chemical functionalities.
These design principles were not merely academic exercises. They directly enabled the creation of peptide drugs with clinically useful pharmacokinetic profiles. The natural GLP-1 peptide, for example, has a plasma half-life of less than 2 minutes. Without the acylation and amino acid modification strategies developed during this era, long-acting GLP-1 agonists like semaglutide (half-life of approximately 7 days) would never have been possible.
The Cadaver Growth Hormone Tragedy
Between 1958 and 1985, approximately 7,700 children in the United States received cadaver-derived growth hormone through the NHPP. Of these, at least 29 developed Creutzfeldt-Jakob disease, a fatal degenerative brain condition caused by prion contamination. Worldwide, over 200 cases of iatrogenic CJD were linked to cadaver growth hormone. The tragedy underscored the urgent need for recombinant alternatives and accelerated the transition to biosynthetic peptide hormones.
The Peptide Drug Industry Takes Shape
The 1990s saw the peptide therapeutics industry consolidate and professionalize. Specialized contract development and manufacturing organizations (CDMOs) emerged to serve pharmaceutical companies that needed peptide synthesis capabilities but didn't want to build them in-house. Companies like Bachem (founded in 1971 in Switzerland), PolyPeptide Group, and Lonza became major suppliers of GMP-grade peptides for clinical trials and commercial production.
The business model for peptide drugs also evolved. In the 1980s, most peptide drugs were produced by large pharmaceutical companies (Eli Lilly, Novo Nordisk, Sanofi) that had vertically integrated manufacturing capabilities. By the 1990s, a more diverse ecosystem had emerged, with biotech startups discovering new peptide drug candidates and licensing them to larger companies for development and commercialization. Amylin Pharmaceuticals, which developed exenatide (Byetta), exemplified this model: a small biotech with a promising molecule partnering with a large pharma company (Eli Lilly) for clinical development, regulatory approval, and marketing.
The 1990s also saw important advances in peptide formulation and delivery. The development of sustained-release microsphere formulations (using biodegradable polymers like PLGA) enabled monthly or quarterly dosing of peptide drugs that would otherwise require daily injections. Lupron Depot (leuprolide acetate for depot suspension), approved in various formulations throughout the 1990s, demonstrated that sophisticated formulation technology could transform the patient experience for peptide drugs. Instead of daily injections, patients could receive an injection once a month, once every three months, or even once every six months.
Nasal and pulmonary delivery routes were also explored during this period. Nasal calcitonin (Miacalcin nasal spray) was approved in 1995 for osteoporosis, demonstrating that some peptides could be delivered across mucosal surfaces. Desmopressin nasal spray for diabetes insipidus had been available even earlier. However, the bioavailability of nasally administered peptides was generally low (1-5%), limiting this route to peptides that are active at very low systemic doses.
Perhaps most significantly for the field's future, the 1990s saw the first serious attempts at oral peptide delivery. While no oral peptide drugs were approved during this decade, the foundational research on absorption enhancers, enteric coatings, protease inhibitors, and mucoadhesive formulations that would eventually enable oral semaglutide (Rybelsus, 2019) was being conducted. The Danish company Emisphere Technologies developed the SNAC (salcaprozate sodium) absorption enhancer technology that Novo Nordisk would later license for oral semaglutide.
The Rise of Peptide Diagnostics
Peptides have become essential not only as therapeutics but also as diagnostic tools. The development of enzyme-linked immunosorbent assays (ELISAs) and radioimmunoassays for peptide hormones transformed clinical endocrinology. Measuring insulin, C-peptide (a fragment released during insulin processing), glucagon, GLP-1, GIP, growth hormone, cortisol, and other peptide hormones became routine clinical practice, enabling precise diagnosis and monitoring of endocrine disorders.
BNP (B-type natriuretic peptide) and NT-proBNP measurements became standard biomarkers for heart failure diagnosis and management, as mentioned earlier. Procalcitonin, a peptide precursor of calcitonin, emerged as a biomarker for bacterial infections and sepsis, helping clinicians distinguish bacterial from viral infections and guide antibiotic therapy decisions.
Peptide-based imaging agents represent another diagnostic application. Radiolabeled somatostatin analogs (like 68Ga-DOTATATE for PET/CT scanning) are used to detect and localize neuroendocrine tumors. PSMA-targeting peptides are used in prostate cancer imaging. These diagnostic peptides often pair with their therapeutic counterparts: the same peptide that localizes a tumor for imaging can be tagged with a therapeutic radioisotope (like Lutetium-177) to treat it. This "theranostic" approach, where the same molecular target is used for both diagnosis and therapy, represents a compelling model for precision medicine.
George Smith, Phage Display, and Peptide Library Screening
In 1985, George P. Smith at the University of Missouri demonstrated that foreign peptide sequences could be displayed on the surface of bacteriophage (bacterial viruses) by fusing them to a phage coat protein. This technique, called phage display, made it possible to create enormous libraries of random peptide sequences (billions of variants) and then screen them for binding to a target protein.
Phage display was further developed by Sir Gregory Winter at the MRC Laboratory of Molecular Biology in Cambridge, who used it to evolve antibodies with desired binding properties. The technique led to the development of adalimumab (Humira), the first fully human monoclonal antibody approved by the FDA (2002), which became the best-selling drug in history.
Smith and Winter shared the 2018 Nobel Prize in Chemistry for their work on phage display and directed evolution of peptides and antibodies. While phage display is most commonly associated with antibody development, it has also been used extensively to discover peptide ligands for drug targets, peptide-based imaging agents, and peptide-targeted drug delivery systems.

2000-2015: GLP-1 Emergence
The incretin story is one of the most remarkable in modern pharmacology. A gut hormone discovered in the 1980s, dismissed by many as therapeutically impractical because of its ultrashort half-life, became the foundation of a drug class that now generates tens of billions of dollars in annual revenue and has changed how physicians treat both diabetes and obesity.
The Incretin Concept: Gut Hormones and Insulin Secretion
The idea that the gut communicates with the pancreas to regulate insulin secretion dates back to the early 20th century. In 1902, Ernest Starling and William Bayliss discovered secretin, the first hormone ever described, and proposed that the intestine released chemical messengers in response to food. In 1932, Belgian physiologist Jean La Barre coined the term "incretin" for a substance from the gut that lowered blood glucose by enhancing insulin release.
The incretin concept was substantiated in the 1960s when researchers showed that oral glucose provoked a much larger insulin response than an equivalent amount of glucose given intravenously. This "incretin effect" accounted for 50-70% of the insulin response to an oral glucose load. Two hormones were eventually identified as the primary mediators: glucose-dependent insulinotropic polypeptide (GIP, originally called gastric inhibitory polypeptide) identified in 1970 by John Brown and colleagues, and glucagon-like peptide-1 (GLP-1).
The Discovery of GLP-1
In 1983, Joel Habener's laboratory at Massachusetts General Hospital cloned and sequenced the proglucagon gene, which encodes the precursor protein for glucagon. They found something unexpected: the gene also encoded two additional glucagon-like sequences, which they named glucagon-like peptide-1 (GLP-1) and glucagon-like peptide-2 (GLP-2).
The initial assumption was that GLP-1, like glucagon, acted primarily on the pancreas. But in 1986 and 1987, several groups independently demonstrated that a truncated form of GLP-1 (specifically, GLP-1(7-36)amide) was a potent stimulator of insulin secretion. Svetlana Mojsov at Massachusetts General Hospital showed that this truncated form, not the full-length GLP-1(1-37), was the biologically active incretin. Jens Juul Holst's group at the University of Copenhagen confirmed these findings and showed that GLP-1 was released from intestinal L-cells in response to meal ingestion.
Daniel Drucker at the University of Toronto made additional contributions, demonstrating GLP-1's trophic effects on pancreatic beta cells and its role in suppressing glucagon secretion. Working together and independently, Habener, Mojsov, Holst, and Drucker built the case that GLP-1 was a major physiological regulator of blood glucose.
The key properties of GLP-1 that made it attractive as a diabetes therapy were:
- Glucose-dependent insulin secretion: GLP-1 stimulates insulin release only when blood glucose is elevated, dramatically reducing the risk of hypoglycemia compared to sulfonylureas or exogenous insulin.
- Glucagon suppression: GLP-1 inhibits glucagon secretion from pancreatic alpha cells, reducing hepatic glucose output.
- Delayed gastric emptying: GLP-1 slows the rate at which food moves from the stomach to the small intestine, reducing postprandial glucose spikes.
- Appetite suppression: GLP-1 acts on receptors in the hypothalamus and brainstem to reduce hunger and promote satiety.
- Beta-cell preservation: In animal models, GLP-1 promoted beta-cell proliferation and inhibited beta-cell apoptosis, raising the possibility that GLP-1-based therapies might slow the progression of type 2 diabetes.
The Half-Life Problem
Despite these attractive properties, GLP-1 had one enormous pharmacological limitation: its plasma half-life was less than 2 minutes. The enzyme dipeptidyl peptidase-4 (DPP-4) rapidly cleaved the first two amino acids from the N-terminus of GLP-1, inactivating it almost immediately after release. Carolyn Deacon and Jens Juul Holst at Copenhagen were instrumental in identifying DPP-4 as the culprit.
This ultrashort half-life meant that continuous intravenous infusion was the only way to maintain therapeutic GLP-1 levels, which was obviously impractical for treating millions of patients with type 2 diabetes. Two strategies emerged to solve this problem:
- DPP-4 inhibitors: Small-molecule drugs that block the enzyme that degrades GLP-1, thereby extending the action of the body's own GLP-1. Sitagliptin (Januvia) became the first DPP-4 inhibitor approved by the FDA in 2006.
- GLP-1 receptor agonists: Modified versions of GLP-1 (or peptides from other species that activate the GLP-1 receptor) that resist DPP-4 cleavage and have extended half-lives.
Exendin-4 and the Gila Monster Connection
The story of how a venomous lizard contributed to diabetes treatment is one of medicine's great yarns. In 1992, John Eng, an endocrinologist at the Veterans Affairs Medical Center in the Bronx, was studying the venoms of various animals looking for bioactive peptides. In the venom of the Gila monster (Heloderma suspectum), a large venomous lizard native to the American Southwest, he found a 39-amino-acid peptide that shared about 53% sequence similarity with human GLP-1.
Eng named this peptide exendin-4. It bound to and activated the human GLP-1 receptor, stimulated insulin secretion, and - critically - was resistant to DPP-4 degradation. Its plasma half-life was approximately 2.4 hours, vastly longer than the less-than-2-minute half-life of native GLP-1. The reason for the DPP-4 resistance was straightforward: exendin-4 has glycine at position 2 rather than alanine, and DPP-4 can't cleave the Gly-2 bond efficiently.
Eng patented exendin-4 in 1995 and licensed it to Amylin Pharmaceuticals, a small biotech company in San Diego. Amylin partnered with Eli Lilly to develop a synthetic version of exendin-4, which they named exenatide.
Exenatide (Byetta): The First GLP-1 Drug
On April 28, 2005, the FDA approved exenatide (Byetta) for the treatment of type 2 diabetes. It was the first GLP-1 receptor agonist to reach the market, and it validated the entire incretin-based therapeutic strategy. Byetta was administered as a twice-daily subcutaneous injection, and clinical trials showed it reduced HbA1c by approximately 0.8-1.0% while also producing modest weight loss (about 2-3 kg).
Byetta's limitations were also apparent. Twice-daily injections were inconvenient, gastrointestinal side effects (especially nausea) were common during initiation, and the weight loss was relatively modest. But the proof of concept was established: targeting the GLP-1 receptor was a viable and safe approach to treating type 2 diabetes.
An extended-release formulation of exenatide (Bydureon), given as a once-weekly injection using microsphere technology, was approved in 2012.
Liraglutide (Victoza/Saxenda): The Acylation Strategy
While Amylin was developing exenatide from lizard venom, Novo Nordisk was taking a different approach. Rather than using a non-human peptide, they started with the human GLP-1(7-37) sequence and engineered it for longer duration of action. The key modification was fatty acid acylation: attaching a C16 palmitoyl fatty acid chain to the lysine at position 26 via a gamma-glutamic acid linker.
This acylation allowed the modified peptide, named liraglutide, to bind non-covalently to serum albumin in the bloodstream. Since albumin has a half-life of about three weeks, the bound liraglutide was protected from both DPP-4 cleavage and renal filtration. The result was a half-life of about 13 hours, enabling once-daily dosing.
Liraglutide was approved by the FDA in January 2010 under the brand name Victoza for type 2 diabetes. Clinical trials showed it reduced HbA1c by 1.0-1.5% and produced weight loss of approximately 2-3 kg at the diabetes dose (1.8 mg). Then came the weight management studies. The SCALE clinical trial program tested liraglutide at a higher dose (3.0 mg daily) specifically for weight loss. The results showed an average weight reduction of about 8% of body weight at 56 weeks, significantly more than placebo. In December 2014, the FDA approved liraglutide 3.0 mg as Saxenda for chronic weight management, making it the first GLP-1 agonist approved specifically for obesity treatment.
The LEADER cardiovascular outcomes trial, published in 2016, demonstrated that liraglutide reduced major adverse cardiovascular events (MACE) by 13% compared to placebo in patients with type 2 diabetes and high cardiovascular risk. This was the first GLP-1 agonist to show cardiovascular benefit, adding a compelling reason beyond glucose control to prescribe the drug.
DPP-4 Inhibitors: The Oral Incretin Strategy
While GLP-1 receptor agonists were being developed as injectable therapies, another approach to exploiting the incretin system emerged: DPP-4 inhibitors. These small-molecule drugs block dipeptidyl peptidase-4, the enzyme responsible for degrading native GLP-1 within minutes of its release. By inhibiting DPP-4, these drugs approximately double circulating GLP-1 levels after meals, enhancing the body's own incretin effect without requiring injection of exogenous peptide.
Sitagliptin (Januvia), developed by Merck, became the first DPP-4 inhibitor approved by the FDA in October 2006. It was followed by saxagliptin (Onglyza, 2009), linagliptin (Tradjenta, 2011), and alogliptin (Nesina, 2013). The DPP-4 inhibitor class offered several practical advantages over early GLP-1 agonists: oral dosing, once-daily administration, minimal gastrointestinal side effects, and a low risk of hypoglycemia.
However, DPP-4 inhibitors produce considerably less weight loss and smaller HbA1c reductions than GLP-1 receptor agonists. The reason is pharmacological: DPP-4 inhibitors can only amplify the body's own GLP-1 production, which is limited by the capacity of intestinal L-cells. GLP-1 receptor agonists, by contrast, can achieve supraphysiological receptor stimulation, producing more pronounced effects on appetite, gastric emptying, and insulin secretion. The clinical consequence is that DPP-4 inhibitors reduce HbA1c by about 0.5-0.8% and produce minimal weight loss (typically 0-1 kg), while GLP-1 agonists reduce HbA1c by 1.0-2.3% and produce weight loss of 2-15+ kg depending on the specific agent and dose.
DPP-4 inhibitors have remained an important part of the diabetes pharmacopeia, particularly for patients who prefer oral medications and don't need aggressive weight management. But the dramatic efficacy differences between DPP-4 inhibitors and GLP-1 agonists have increasingly shifted prescribing patterns toward the latter, especially as injectable GLP-1 agonists have become available in convenient once-weekly formulations.
The Broader Incretin Biology
Research into incretin biology during this period revealed that GLP-1's effects extended far beyond the pancreas. GLP-1 receptors were identified in the heart, blood vessels, kidneys, brain, liver, and immune cells, suggesting a broader physiological role than initially appreciated. Studies showed that GLP-1 reduced hepatic glucose production independently of insulin, improved myocardial function in animal models of heart failure, reduced neuroinflammation, and modulated immune responses.
These pleiotropic effects have become increasingly relevant as GLP-1 agonists are tested for indications beyond diabetes. The cardiovascular benefits observed in the LEADER, SUSTAIN-6, and SELECT trials may reflect direct cardioprotective effects of GLP-1 receptor activation, not just secondary benefits of improved glycemic control and weight loss. Similarly, the neuroprotective effects observed in preclinical studies are driving interest in GLP-1 agonists for Alzheimer's and Parkinson's disease.
GLP-2, the "other" peptide encoded by the proglucagon gene alongside GLP-1, has also proven therapeutically useful. Unlike GLP-1, GLP-2 acts primarily on the intestinal epithelium, promoting growth and repair of the intestinal lining. Teduglutide (Gattex), a DPP-4-resistant analog of GLP-2 developed based on Daniel Drucker's research, was approved in 2012 for short bowel syndrome. It reduces the need for parenteral nutrition by enhancing intestinal absorptive capacity, demonstrating that the proglucagon-derived peptide family has therapeutic potential beyond metabolic disease.
The incretin system has also intersected with the amylin system. Amylin, a 37-amino-acid peptide co-secreted with insulin from pancreatic beta cells, contributes to glucose regulation by slowing gastric emptying, suppressing glucagon secretion, and reducing food intake. Pramlintide (Symlin), a synthetic amylin analog, was approved in 2005 for diabetes. Cagrilintide, Novo Nordisk's long-acting amylin analog, is now being combined with semaglutide in CagriSema to target both incretin and amylin pathways simultaneously, reflecting the broader trend toward multi-pathway metabolic therapy.
The Competitive Landscape: 2005-2015
The decade following exenatide's approval saw a rapid proliferation of GLP-1 receptor agonists and DPP-4 inhibitors:
| Drug | Brand | FDA Approval | Dosing | Type |
|---|---|---|---|---|
| Exenatide | Byetta | April 2005 | Twice daily | GLP-1 agonist (exendin-4) |
| Sitagliptin | Januvia | October 2006 | Once daily (oral) | DPP-4 inhibitor |
| Liraglutide | Victoza | January 2010 | Once daily | GLP-1 agonist (acylated) |
| Exenatide ER | Bydureon | January 2012 | Once weekly | GLP-1 agonist (microsphere) |
| Albiglutide | Tanzeum | April 2014 | Once weekly | GLP-1 agonist (albumin fusion) |
| Liraglutide 3mg | Saxenda | December 2014 | Once daily | GLP-1 agonist (weight loss) |
| Dulaglutide | Trulicity | September 2014 | Once weekly | GLP-1 agonist (Fc fusion) |
Each successive drug refined the pharmacology. The trend was unmistakable: less frequent dosing, better efficacy, fewer side effects, and expanding indications beyond diabetes into obesity and cardiovascular risk reduction. But the biggest leap was still to come.
The Incretin Effect in Practice
GLP-1 receptor agonists exploit a natural physiological system. After a meal, intestinal L-cells release GLP-1, which stimulates insulin secretion, suppresses glucagon, slows gastric emptying, and reduces appetite. In type 2 diabetes, this incretin response is blunted. GLP-1 agonists restore and amplify the incretin effect, and because insulin stimulation is glucose-dependent, the risk of hypoglycemia is far lower than with sulfonylureas or insulin. This safety profile was crucial for the widespread adoption of GLP-1 therapies.
2015-Present: The GLP-1 Transformation
If the decade from 2005 to 2015 was about proving the GLP-1 concept, the period from 2015 to 2024 has been about scaling it into one of the most impactful drug classes in pharmaceutical history. Semaglutide and tirzepatide have delivered unprecedented weight loss results, earned cardiovascular indications, and captured the public imagination in ways that few prescription drugs ever have.
Semaglutide: Novo Nordisk's Masterwork
Semaglutide represents the culmination of Novo Nordisk's decades of GLP-1 expertise. Like liraglutide, semaglutide is based on a modified human GLP-1 sequence. But the engineering is more sophisticated. Three key modifications give semaglutide its exceptional pharmacological profile:
- Amino acid substitution at position 8: Alanine is replaced with alpha-aminobutyric acid (Aib), which confers near-complete resistance to DPP-4 cleavage.
- Fatty diacid acylation at position 26: A C18 fatty diacid chain is attached via a linker to the lysine at position 26. This provides strong, reversible binding to serum albumin, dramatically extending plasma half-life.
- Amino acid substitution at position 34: Lysine is replaced with arginine to prevent acylation at the wrong position.
The result is a GLP-1 analog with a plasma half-life of approximately 165 hours (roughly 7 days), enabling once-weekly subcutaneous injection. This was a massive improvement over liraglutide's 13-hour half-life and once-daily dosing.
Ozempic: Diabetes Approval (2017)
The FDA approved semaglutide injection (Ozempic) for type 2 diabetes in December 2017. The SUSTAIN clinical trial program demonstrated that Ozempic reduced HbA1c by 1.5-1.8% and produced weight loss of 4.5-6.5 kg (approximately 5-7% of body weight) at the 1.0 mg dose. These were the best efficacy results of any GLP-1 agonist approved up to that point.
Rybelsus: Oral GLP-1 (2019)
In September 2019, the FDA approved oral semaglutide (Rybelsus) for type 2 diabetes, a remarkable technical achievement. Peptide drugs are typically destroyed by stomach acid and digestive enzymes, making oral delivery extremely challenging. Novo Nordisk solved this problem by co-formulating semaglutide with SNAC (sodium N-[8-(2-hydroxybenzoyl)amino]caprylate), an absorption enhancer that promotes transcellular absorption of the peptide across the gastric mucosa.
Oral semaglutide must be taken on an empty stomach with no more than 120 mL of water, and the patient must wait at least 30 minutes before eating, drinking, or taking other medications. Despite these restrictions, the availability of an oral GLP-1 agonist was a significant advance for patients who are averse to injections.
Wegovy: The Weight Loss Phenomenon (2021)
The approval that changed everything came on June 4, 2021, when the FDA approved semaglutide 2.4 mg weekly injection (Wegovy) for chronic weight management. The STEP clinical trial program had produced striking results:
- STEP 1: Adults with obesity (BMI ≥30) or overweight (BMI ≥27) with at least one weight-related comorbidity lost an average of 14.9% of their body weight over 68 weeks with semaglutide 2.4 mg versus 2.4% with placebo.
- STEP 2: Adults with type 2 diabetes and obesity lost an average of 9.6% of body weight.
- STEP 3: Combined with intensive behavioral therapy, participants lost an average of 16.0% of body weight.
- STEP 5: Extended 104-week data showed sustained weight loss of approximately 15.2%.
These results were unprecedented for a pharmacological intervention. Previous anti-obesity drugs typically produced 5-10% weight loss. Semaglutide 2.4 mg nearly doubled or tripled those results. The medical community took notice, and so did the general public. Ozempic and Wegovy became household names, featured on magazine covers, discussed on social media, and debated in op-ed columns.
SELECT Trial: Cardiovascular Protection (2023)
In August 2023, the results of the SELECT cardiovascular outcomes trial were announced. SELECT enrolled over 17,600 adults with overweight or obesity and established cardiovascular disease (but without diabetes). Semaglutide 2.4 mg reduced the risk of major adverse cardiovascular events (MACE) by 20% compared to placebo over a median follow-up of approximately 40 months. This was the first time a weight-loss drug had demonstrated cardiovascular benefit in people without diabetes, and it led to an expanded FDA indication for Wegovy in March 2024.
Tirzepatide: The Dual Agonist
While Novo Nordisk was perfecting semaglutide, Eli Lilly was developing tirzepatide, a fundamentally different molecule. Tirzepatide is a dual agonist that activates both the GLP-1 receptor and the GIP (glucose-dependent insulinotropic polypeptide) receptor. It's a 39-amino-acid linear peptide with a C20 fatty diacid moiety that enables once-weekly dosing.
The rationale for dual agonism was based on the observation that both GLP-1 and GIP contribute to the incretin effect, but they act through different receptors and partially distinct signaling pathways. By activating both receptors, tirzepatide might produce additive or complementary effects on glucose control, weight loss, and metabolic health.
Mounjaro: Diabetes Approval (2022)
The FDA approved tirzepatide (Mounjaro) for type 2 diabetes in May 2022. The SURPASS trial program showed remarkable efficacy:
- HbA1c reductions of 2.0-2.3% (the largest of any GLP-1-class drug)
- Weight loss of 7-13 kg (7-13% body weight) at the diabetes doses
- In the SURPASS-2 head-to-head trial against semaglutide 1.0 mg, tirzepatide at all three doses (5 mg, 10 mg, 15 mg) produced significantly greater HbA1c reduction and weight loss
Zepbound: Weight Loss Approval (2023)
In November 2023, the FDA approved tirzepatide as Zepbound for chronic weight management. The SURMOUNT-1 trial results were extraordinary: at the 15 mg dose, participants lost an average of 22.5% of their body weight over 72 weeks. At the 10 mg dose, weight loss averaged 21.4%. Even the lowest dose (5 mg) produced 16.0% weight loss. These results exceeded those of semaglutide 2.4 mg and established tirzepatide as the most effective approved anti-obesity medication.
The Pipeline: What's Coming Next
The success of semaglutide and tirzepatide has triggered an avalanche of GLP-1-related drug development. As of 2024, the pipeline includes:
- Retatrutide (Eli Lilly): A triple agonist targeting GLP-1, GIP, and glucagon receptors. Phase 2 data showed weight loss up to 24.2% at 48 weeks, the highest ever reported for a pharmacological agent. Phase 3 trials are ongoing.
- Orforglipron (Eli Lilly): A small-molecule, non-peptide oral GLP-1 agonist. Unlike oral semaglutide, orforglipron doesn't require an absorption enhancer and can be taken with food. Phase 3 trials are underway.
- Survodutide (Boehringer Ingelheim/Zealand): A dual GLP-1/glucagon agonist being developed for obesity and NASH/MASH.
- CagriSema (Novo Nordisk): A combination of semaglutide with cagrilintide (a long-acting amylin analog). Phase 3 data showed weight loss of approximately 22-24%.
- Pemvidutide (Altimmune): A dual GLP-1/glucagon agonist in Phase 2 for obesity and NASH.
The GLP-1 class continues to expand in scope. Research is exploring these drugs for non-alcoholic fatty liver disease (NAFLD/MASH), heart failure with preserved ejection fraction, chronic kidney disease, Alzheimer's disease, addiction, and sleep apnea. Each positive trial result widens the potential patient population and strengthens the commercial case.
FDA Peptide Drug Approvals by Decade
| Drug | Brand | Approval | Max Weight Loss | Mechanism |
|---|---|---|---|---|
| Semaglutide 2.4mg | Wegovy | June 2021 | ~15% (68 weeks) | GLP-1 agonist |
| Tirzepatide 15mg | Zepbound | November 2023 | ~22.5% (72 weeks) | GLP-1/GIP dual agonist |
| Retatrutide 12mg | TBD (Phase 3) | Expected 2026-2027 | ~24.2% (48 weeks, Phase 2) | GLP-1/GIP/Glucagon triple agonist |
Key Figures in Peptide Medicine
The history of peptide medicine has been shaped by a remarkable collection of scientists, clinicians, and entrepreneurs. Some won Nobel Prizes. Others worked in relative obscurity for decades before their contributions were recognized. All of them pushed the boundaries of what was possible.
Frederick Banting (1891-1941)
A Canadian surgeon with no research training who had the insight that ligating pancreatic ducts could preserve the islets of Langerhans. His persistence in pursuing this idea, despite skepticism from the established scientific community, led to the isolation of insulin. At 32, he became the youngest Nobel laureate in Physiology or Medicine at that time. He shared his prize money with Charles Best. Banting was killed in a plane crash in Newfoundland in 1941 while traveling on a wartime medical mission.
Charles Best (1899-1978)
The medical student who won a coin toss and ended up as Banting's assistant during the summer of 1921. Best performed most of the hands-on experimental work in the early insulin experiments, including the blood sugar measurements. He was controversially excluded from the Nobel Prize, but Banting insisted on sharing his prize money with Best. Best later became head of the Department of Physiology at the University of Toronto and continued to contribute to diabetes research for decades.
Frederick Sanger (1918-2013)
A British biochemist who won two Nobel Prizes in Chemistry, a feat achieved by no other person. His first Nobel (1958) was for determining the amino acid sequence of insulin, establishing that proteins have definite sequences. His second Nobel (1980) was for developing methods for DNA sequencing. Both achievements were foundational for molecular biology and peptide drug development.
Vincent du Vigneaud (1901-1978)
An American biochemist whose lifelong fascination with sulfur chemistry led him to synthesize oxytocin, the first peptide hormone ever created from scratch. His 1955 Nobel Prize in Chemistry recognized the broader significance of this work: it proved that biological activity could be reproduced by chemical synthesis, opening the door to synthetic peptide drugs.
R. Bruce Merrifield (1921-2006)
The inventor of solid-phase peptide synthesis, arguably the single most important enabling technology in the history of peptide drug development. Merrifield's 1963 paper transformed peptide chemistry from an artisanal craft into an automated, reproducible process. He received the Nobel Prize in Chemistry in 1984. Born in Fort Worth, Texas, he spent his entire career at The Rockefeller Institute/University in New York.
Roger Guillemin (1924-2024) and Andrew Schally (1926-present)
The bitter rivals who spent decades isolating hypothalamic releasing hormones, processing millions of animal hypothalami in the process. Their discoveries of TRH, GnRH, somatostatin, and GHRH opened up neuroendocrinology as a field and led directly to multiple peptide drugs. They shared the 1977 Nobel Prize in Physiology or Medicine with Rosalyn Yalow. Despite their rivalry, both scientists' work was complementary and mutually reinforcing.
Rosalyn Yalow (1921-2011)
Co-developer (with Solomon Berson) of radioimmunoassay, the technique that made it possible to measure peptide hormones in blood. Without RIA, the discoveries of Guillemin, Schally, and many others would have been impossible. Yalow was the second woman to win the Nobel Prize in Physiology or Medicine and the first American-born woman to do so.
Jens Juul Holst (1945-present)
A Danish physician-scientist at the University of Copenhagen whose five decades of work on GLP-1 physiology laid the foundation for the entire GLP-1 drug class. Holst's group demonstrated that GLP-1(7-36)amide was the biologically active form of GLP-1, characterized its actions on insulin secretion and appetite, and identified DPP-4 as the enzyme responsible for its rapid degradation. He received the Lasker-DeBakey Clinical Medical Research Award in 2024, along with Joel Habener and Svetlana Mojsov.
Joel Habener (1937-present)
An American endocrinologist at Massachusetts General Hospital and Harvard Medical School who cloned the proglucagon gene and discovered the GLP-1 and GLP-2 peptides encoded within it. His work provided the molecular foundation for the entire incretin therapy field. Co-recipient of the 2024 Lasker Award.
Svetlana Mojsov (1951-present)
A peptide chemist at Massachusetts General Hospital and later The Rockefeller University who synthesized the truncated, amidated form of GLP-1 that proved to be the biologically active incretin. Her careful synthetic chemistry and bioassay work was essential for identifying GLP-1(7-36)amide as the potent insulinotropic peptide. Co-recipient of the 2024 Lasker Award.
Daniel Drucker (1956-present)
A Canadian endocrinologist at the University of Toronto and Mount Sinai Hospital who characterized GLP-1's effects on beta-cell biology, glucagon suppression, and gut physiology. Drucker's laboratory demonstrated GLP-2's trophic effects on the intestinal epithelium, leading to the development of teduglutide (Gattex) for short bowel syndrome. He received the Canada Gairdner International Award in 2021.
John Eng (1953-present)
The endocrinologist who discovered exendin-4 in Gila monster venom in 1992, leading directly to the development of exenatide (Byetta), the first GLP-1 receptor agonist approved for clinical use. Eng's persistence in studying lizard venom peptides, initially met with skepticism, ultimately validated a novel approach to drug discovery from natural sources.

Nobel Prizes in Peptide Research
Peptide science has generated an extraordinary number of Nobel Prizes. At least twelve Nobel awards can be directly connected to peptide discovery, synthesis, analysis, or application. This concentration of Nobel-level work in a single field reflects both the fundamental importance of peptides in biology and the technical challenges that had to be overcome to study them.
| Year | Prize | Laureate(s) | Contribution |
|---|---|---|---|
| 1923 | Physiology or Medicine | Frederick Banting, J.J.R. Macleod | Discovery of insulin |
| 1955 | Chemistry | Vincent du Vigneaud | First synthesis of a polypeptide hormone (oxytocin) |
| 1958 | Chemistry | Frederick Sanger | Determination of the amino acid sequence of insulin |
| 1964 | Chemistry | Dorothy Crowfoot Hodgkin | X-ray crystallographic determination of insulin and other biochemical structures |
| 1972 | Chemistry | Christian Anfinsen, Stanford Moore, William Stein | Studies on ribonuclease, amino acid analysis of proteins |
| 1977 | Physiology or Medicine | Roger Guillemin, Andrew Schally, Rosalyn Yalow | Hypothalamic peptide hormones and radioimmunoassay |
| 1980 | Chemistry | Frederick Sanger (2nd), Walter Gilbert, Paul Berg | DNA sequencing and recombinant DNA (enabling recombinant insulin) |
| 1984 | Chemistry | R. Bruce Merrifield | Solid-phase peptide synthesis |
| 2004 | Chemistry | Aaron Ciechanover, Avram Hershko, Irwin Rose | Discovery of ubiquitin-mediated protein degradation |
| 2012 | Chemistry | Robert Lefkowitz, Brian Kobilka | G protein-coupled receptors (the receptor family that includes the GLP-1 receptor) |
| 2018 | Chemistry | George Smith, Gregory Winter, Frances Arnold | Phage display of peptides and directed evolution |
| 2024 | Chemistry | David Baker, Demis Hassabis, John Jumper | Computational protein structure prediction (AlphaFold) and protein design |
The Significance of This Nobel Legacy
Several observations emerge from this list. First, peptide-related Nobel Prizes span the full range of the natural sciences. They've been awarded in both Chemistry and Physiology or Medicine, reflecting peptides' dual nature as both chemical entities and biological agents. Second, the prizes cluster around enabling technologies - sequencing, synthesis, assay methods, structural determination - that opened up entire fields of investigation. Third, the most recent prizes (2018, 2024) point toward the future: phage display, directed evolution, and computational protein design are the tools that will drive the next generation of peptide drug discovery.
The 2024 Chemistry Nobel for AlphaFold (Hassabis and Jumper) and computational protein design (Baker) is particularly relevant to peptide medicine's future. AlphaFold can predict the three-dimensional structure of virtually any protein, including peptide hormone receptors, with near-experimental accuracy. Baker's laboratory has designed entirely novel peptide and protein structures that don't exist in nature, opening the possibility of engineering therapeutic peptides with properties unachievable through modification of natural sequences. These computational tools are expected to dramatically accelerate peptide drug discovery in the coming decade.
The 2012 Chemistry Nobel for G protein-coupled receptors (GPCRs) also deserves mention. Robert Lefkowitz and Brian Kobilka's work on the structure and function of GPCRs provided the molecular framework for understanding how peptide hormones like GLP-1 activate their receptors. The crystal and cryo-EM structures of the GLP-1 receptor in complex with various agonists have been essential for designing new GLP-1 drugs with optimized receptor binding and signaling properties.
A Nobel-Dense Field
It's difficult to think of another area of medicine that has generated as many Nobel Prizes as peptide science. The concentration of Nobel-level work reflects the fact that peptides sit at the intersection of chemistry, biology, and medicine. Understanding them required advances in organic chemistry (synthesis), analytical chemistry (sequencing, assays), structural biology (crystallography, NMR), molecular biology (gene cloning), and pharmacology (receptor biology). Each breakthrough opened new questions and new opportunities.
The Evolution of Peptide Synthesis Technology
The ability to synthesize peptides has progressed from a painstaking, artisanal endeavor requiring years of effort for a single molecule to an automated, industrialized process capable of producing tons of pharmaceutical-grade peptide per year. This evolution in manufacturing technology has been as important as biological discovery in making peptide medicine a reality.
Classical Solution-Phase Synthesis (1901-1962)
The very first peptide synthesis was achieved by Emil Fischer in 1901, when he coupled glycine to glycine using an acid chloride method. Fischer went on to synthesize several short peptides and coined the term "peptide" itself. His work demonstrated that amino acids could be linked by chemical bonds to form chains, but the methods were crude and yields were low.
For the next six decades, peptide chemists worked in solution, building peptide chains one amino acid at a time. Each coupling required careful protection of all reactive groups except the ones intended to react, followed by activation of the carboxyl group and coupling, then removal of the protecting group for the next cycle. Each step required purification of the product, typically by crystallization or chromatography. Yields for each coupling step ranged from 60% to 90%, which meant that for a 10-residue peptide, overall yields could be catastrophically low (as little as 1-10% for longer sequences).
The protecting group chemistry of this era was limited. The carbobenzoxy (Cbz or Z) group, introduced by Max Bergmann and Leonidas Zervas in 1932, was the first widely used N-terminal protecting group. It could be removed by hydrogenolysis (catalytic reduction with hydrogen) without affecting peptide bonds. The tert-butyloxycarbonyl (Boc) group, developed by Louis Carpino and G.Y. Han in 1957, offered an orthogonal protection strategy: it could be removed by mild acid treatment. These protecting groups were essential tools for building peptides, but solution-phase synthesis remained slow and labor-intensive.
Despite these challenges, solution-phase synthesis produced several landmark achievements. Besides du Vigneaud's synthesis of oxytocin (1953) and vasopressin, Panayotis Katsoyannis at the Brookhaven National Laboratory and a Chinese team led by Niu Jingyi independently reported the total synthesis of insulin in the early 1960s. These were heroic efforts involving dozens of chemists working for several years each. The insulin syntheses, completed around 1963-1965, demonstrated that even complex peptide hormones could be built from scratch, but they also illustrated the practical limitations of solution-phase methods.
The Merrifield Revolution: Solid-Phase Peptide Synthesis (1963-1980s)
Merrifield's 1963 paper on SPPS didn't just improve yields and speed. It changed the entire intellectual framework of peptide chemistry. Before SPPS, each peptide synthesis was a unique research project. After SPPS, synthesis became a systematic, repetitive process that could be optimized and automated.
The original Merrifield method used Boc (tert-butyloxycarbonyl) chemistry for N-terminal protection. The peptide was built from the C-terminus, with each amino acid added in the N-to-C direction. After coupling, the Boc group was removed with trifluoroacetic acid (TFA), and the next amino acid was coupled. Side-chain protecting groups were chosen to be stable under the Boc-removal conditions. At the end of the synthesis, the peptide was cleaved from the resin and all remaining protecting groups were removed simultaneously using strong acid (typically hydrogen fluoride, HF).
The use of anhydrous HF was one of the less convenient aspects of Boc SPPS. HF is extremely corrosive and toxic, requiring specialized equipment and careful handling. This limitation motivated the development of alternative approaches.
Fmoc Chemistry: A Gentler Alternative
In 1970, Louis Carpino introduced the 9-fluorenylmethyloxycarbonyl (Fmoc) group as an alternative to Boc. The Fmoc group is removed by mild base (typically 20% piperidine in dimethylformamide) rather than acid. This orthogonality meant that acid-labile side-chain protecting groups (like tert-butyl esters and ethers) could be used alongside Fmoc, and the final cleavage from resin and side-chain deprotection could be accomplished with TFA rather than HF.
Fmoc-based SPPS, refined and popularized through the 1980s and 1990s by Atherton, Sheppard, Fields, and others, eventually became the dominant method for peptide synthesis. Its advantages included milder reaction conditions, better compatibility with sensitive amino acids (like tryptophan, methionine, and cysteine), and greater amenability to automation. By the 1990s, automated peptide synthesizers using Fmoc chemistry could produce a 30-residue peptide overnight.
Automation and the First Peptide Synthesizers
Merrifield himself built the first automated peptide synthesizer in the 1960s, a custom machine that performed the repetitive cycles of coupling, washing, and deprotection without manual intervention. By the 1970s, several commercial companies (Beckman, Applied Biosystems, Advanced ChemTech) began selling automated peptide synthesizers. These instruments made SPPS accessible to biology laboratories that lacked specialist peptide chemistry expertise.
The introduction of microwave-assisted SPPS in the 2000s further accelerated synthesis. Microwave irradiation speeds up coupling reactions and reduces aggregation of the growing peptide chain, enabling synthesis of difficult sequences that would fail under standard conditions. Modern microwave peptide synthesizers (such as the CEM Liberty series) can produce a 50-residue peptide in under 4 hours.
Large-Scale Manufacturing: From Lab Bench to Factory Floor
Manufacturing peptides at pharmaceutical scale presents different challenges than laboratory synthesis. The quantities involved are much larger (kilograms to tons per year for drugs like semaglutide), and every batch must meet stringent regulatory standards for purity, consistency, and documentation.
Three main approaches are used for large-scale peptide production:
Chemical Synthesis at Scale
Large-scale SPPS uses the same chemistry as laboratory SPPS but with industrial-sized reactors, optimized reagent recycling, and sophisticated purification processes. For peptides up to about 40 amino acids, chemical synthesis is often the preferred method. Semaglutide, for example, is produced by chemical synthesis. The 31-amino-acid backbone is assembled by SPPS (or by fragment condensation, combining pre-synthesized peptide fragments in solution), and the C18 fatty diacid linker is then attached chemically. Purification is accomplished by reverse-phase high-performance liquid chromatography (RP-HPLC), often requiring multiple chromatographic steps to achieve the required purity (typically >98%).
The cost of large-scale SPPS has decreased dramatically over the past two decades. Improvements in coupling reagents, resin technology, solvent recycling, and purification methods have reduced the cost of goods for synthetic peptides from thousands of dollars per gram to tens of dollars per gram for well-optimized processes.
Recombinant Production
For larger peptides and proteins, recombinant production in bacteria (E. coli), yeast (Pichia pastoris, Saccharomyces cerevisiae), or mammalian cells (CHO cells) is often more economical. Recombinant insulin, growth hormone, and many other peptide drugs are produced this way. The gene encoding the desired peptide is inserted into a host cell, which then expresses the peptide as it grows. The peptide is then purified from the cell culture broth.
Recombinant production works best for peptides that consist entirely of the 20 natural amino acids, since the cellular machinery can only incorporate genetically encoded amino acids. Peptides containing unnatural amino acids, unusual modifications (like the Aib substitution in semaglutide), or non-peptide elements (like fatty acid chains) generally require chemical synthesis or hybrid approaches.
Hybrid Approaches
Many modern peptide drugs are manufactured using a combination of recombinant and chemical methods. For instance, the peptide backbone might be produced recombinantly, and then chemical modifications (acylation, PEGylation, glycosylation) are added in subsequent synthetic steps. This approach combines the cost efficiency of recombinant production with the flexibility of chemical modification.
Analytical Methods: Verifying What You've Built
As synthesis methods improved, so did the analytical tools for characterizing peptides. Mass spectrometry (MS), especially matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI) techniques, made it possible to confirm the molecular weight of synthetic peptides with extraordinary precision. Nuclear magnetic resonance (NMR) spectroscopy provided structural information about peptide conformation in solution. X-ray crystallography and cryo-electron microscopy (cryo-EM) revealed three-dimensional structures at atomic resolution.
Modern quality control for pharmaceutical-grade peptides employs a battery of analytical techniques: RP-HPLC for purity assessment, mass spectrometry for identity confirmation, amino acid analysis for composition verification, peptide mapping (enzymatic digestion followed by LC-MS) for sequence confirmation, and various assays for potency, sterility, and endotoxin content. These methods ensure that every batch of a peptide drug is safe and effective.
The Future of Peptide Synthesis
Several emerging technologies promise to further transform peptide manufacturing:
- Flow chemistry: Continuous-flow synthesis systems can produce peptides more efficiently than batch methods, with better heat transfer, faster reaction times, and reduced solvent waste. The Pentelute laboratory at MIT has demonstrated flow-based SPPS systems that can synthesize a 30-residue peptide in under 40 minutes.
- Enzymatic synthesis: Engineered enzymes (like sortase, butelase, and peptiligase) can catalyze peptide bond formation with high selectivity and under mild conditions. Enzymatic ligation of peptide fragments is being explored as a more sustainable alternative to chemical ligation methods.
- Cell-free protein synthesis: In vitro translation systems can produce peptides using purified ribosomes and translation factors, bypassing the need for living cells. These systems can incorporate unnatural amino acids at specific positions using engineered tRNAs and aminoacyl-tRNA synthetases.
- Green chemistry approaches: Traditional SPPS uses large volumes of organic solvents (DMF, DCM, NMP), many of which are environmentally problematic. Research into water-based coupling reactions, recyclable resins, and biodegradable reagents aims to make peptide synthesis more sustainable.
Key Milestones Timeline: A Century of Peptide Medicine
This comprehensive timeline captures the defining moments in the history of peptide medicine, from pre-insulin discoveries through the modern GLP-1 era. Each entry represents a turning point that moved the field forward.
Pre-Insulin Era (1889-1920)
| Year | Milestone | Significance |
|---|---|---|
| 1889 | Minkowski and von Mering demonstrate that pancreatectomy causes diabetes in dogs | Established the pancreas as the source of a blood-sugar-regulating substance |
| 1901 | Emil Fischer synthesizes the first peptide bond (glycylglycine) | Proved that amino acids could be chemically linked; coined the term "peptide" |
| 1902 | Bayliss and Starling discover secretin, the first hormone | Established the concept of chemical messengers (hormones) controlling organ function |
| 1905 | Starling coins the word "hormone" | Created the conceptual framework for endocrinology and peptide hormone research |
| 1916 | Sharpey-Schafer names the hypothetical pancreatic substance "insulin" | Provided the theoretical target that Banting and Best would pursue five years later |
The Insulin Revolution (1921-1950)
| Year | Milestone | Significance |
|---|---|---|
| 1921 | Banting and Best isolate insulin from canine pancreas | First peptide hormone isolated; proof that an internal secretion controlled blood sugar |
| 1922 | Leonard Thompson receives first insulin injection (January 11) | First therapeutic use of a peptide drug in a human patient |
| 1922 | Eli Lilly begins large-scale insulin production | Established the model for industrial peptide drug manufacturing |
| 1923 | Nobel Prize to Banting and Macleod for insulin | First Nobel Prize directly related to a peptide hormone |
| 1932 | La Barre coins the term "incretin" | Introduced the concept that would eventually lead to GLP-1 drug development |
| 1936 | Hagedorn develops protamine insulin (NPH) | First long-acting insulin formulation; demonstrated that peptide pharmacokinetics could be engineered |
| 1945 | Sanger begins insulin sequencing work | Initiated the first determination of a complete protein amino acid sequence |
Peptide Chemistry Golden Age (1950-1980)
| Year | Milestone | Significance |
|---|---|---|
| 1953 | du Vigneaud synthesizes oxytocin | First total synthesis of a peptide hormone; proved biological activity could be recreated chemically |
| 1955 | Sanger completes insulin amino acid sequence | First complete protein sequence; established that proteins have unique, definite structures |
| 1955 | Nobel Prize to du Vigneaud for oxytocin synthesis | Recognized the broader significance of synthetic peptide chemistry |
| 1958 | Nobel Prize to Sanger for insulin sequencing | Validated the idea that protein structure could be precisely determined |
| 1959 | Yalow and Berson develop radioimmunoassay (RIA) | Made it possible to measure peptide hormones at picomolar concentrations |
| 1963 | Merrifield publishes solid-phase peptide synthesis | Transformed peptide chemistry from artisanal craft to automated science |
| 1964 | Hodgkin determines insulin 3D structure by X-ray crystallography | First three-dimensional structure of a peptide hormone |
| 1969 | TRH structure determined by Schally and Guillemin groups | First hypothalamic releasing hormone characterized; opened neuroendocrinology |
| 1970 | GIP discovered by John Brown | First incretin hormone identified |
| 1971 | GnRH structure determined | Led directly to GnRH analog drugs for cancer, endometriosis, and infertility |
| 1973 | Somatostatin isolated by Guillemin | Led to octreotide and other somatostatin analog drugs |
| 1975 | Enkephalins discovered by Hughes and Kosterlitz | First endogenous opioid peptides; opened the field of opioid peptide biology |
| 1977 | Nobel Prize to Guillemin, Schally, and Yalow | Recognized hypothalamic hormones and radioimmunoassay |
Biotechnology Revolution (1978-2000)
| Year | Milestone | Significance |
|---|---|---|
| 1978 | Recombinant human insulin produced in E. coli | First human protein made by recombinant DNA technology |
| 1982 | Humulin (recombinant insulin) approved by FDA | First recombinant DNA drug; transformed pharmaceutical manufacturing |
| 1983 | Habener clones the proglucagon gene; GLP-1 sequence identified | Discovery of GLP-1, the peptide that would drive a $100 billion drug market |
| 1984 | Nobel Prize to Merrifield for SPPS | Recognized the enabling technology for the entire peptide drug industry |
| 1985 | Recombinant growth hormone (Protropin) approved; cadaver GH recalled | Eliminated risk of prion disease transmission; demonstrated urgency of recombinant alternatives |
| 1985 | George Smith develops phage display | Enabled screening of billions of peptide variants for drug discovery |
| 1986-87 | GLP-1(7-36)amide shown to be a potent insulinotropic hormone | Identified the active form of GLP-1; established it as a therapeutic target |
| 1988 | Octreotide approved for acromegaly | First somatostatin analog drug; demonstrated that peptide analogs could be clinically useful |
| 1989 | Epoetin alfa (Epogen) approved | Became one of the best-selling biologic drugs; demonstrated market potential for peptide drugs |
| 1992 | John Eng discovers exendin-4 in Gila monster venom | Found a natural DPP-4-resistant GLP-1 receptor agonist; led directly to exenatide |
| 1996 | Lispro (Humalog) approved: first insulin analog | Demonstrated that engineering peptide sequence could improve pharmacokinetics |
GLP-1 Era (2005-Present)
| Year | Milestone | Significance |
|---|---|---|
| 2005 | Exenatide (Byetta) approved: first GLP-1 receptor agonist | Validated the incretin therapeutic strategy; proof of concept for GLP-1 drugs |
| 2006 | Sitagliptin (Januvia) approved: first DPP-4 inhibitor | First oral incretin-based therapy; broadened patient access |
| 2010 | Liraglutide (Victoza) approved: first acylated GLP-1 analog | Once-daily dosing through albumin binding; improved convenience and efficacy |
| 2014 | Liraglutide 3.0 mg (Saxenda) approved for weight loss | First GLP-1 agonist approved specifically for obesity treatment |
| 2016 | LEADER trial shows cardiovascular benefit for liraglutide | First GLP-1 agonist to demonstrate cardioprotection |
| 2017 | Semaglutide (Ozempic) approved for diabetes | Once-weekly dosing with superior efficacy; became a blockbuster drug |
| 2019 | Oral semaglutide (Rybelsus) approved | First oral GLP-1 agonist; broke the injection barrier for peptide drugs |
| 2021 | Semaglutide 2.4 mg (Wegovy) approved for weight loss | Produced unprecedented ~15% weight loss; became a cultural phenomenon |
| 2022 | Tirzepatide (Mounjaro) approved for diabetes | First dual GLP-1/GIP agonist; validated multi-receptor approach |
| 2023 | Tirzepatide (Zepbound) approved for weight loss | Most effective approved anti-obesity drug (~22.5% weight loss) |
| 2023 | SELECT trial: semaglutide reduces cardiovascular events in obesity | First weight-loss drug to show CV benefit in people without diabetes |
| 2023 | Retatrutide Phase 2 results: 24.2% weight loss | Triple agonist shows highest weight loss ever reported for a drug |
| 2024 | Nobel Prize for AlphaFold and computational protein design | Signals the beginning of AI-driven peptide drug discovery |
| 2024 | Lasker Award to Habener, Holst, and Mojsov for GLP-1 discovery | Long-overdue recognition of the scientists who founded the GLP-1 field |
The Modern Peptide Therapeutics Explosion
While GLP-1 agonists have captured most of the headlines, the broader peptide therapeutics field is experiencing a period of extraordinary growth and diversification. From antimicrobial peptides to cancer vaccines, peptide-based medicines are finding applications across nearly every area of clinical practice.
Peptide Therapeutics by Clinical Area
Oncology
Peptides are playing an increasingly important role in cancer treatment. Lutetium-177 PSMA (Pluvicto), approved in 2022 for metastatic castration-resistant prostate cancer, uses a peptide ligand to target the prostate-specific membrane antigen (PSMA) on tumor cells, delivering a radioactive payload directly to the cancer. This "radioligand therapy" approach has shown remarkable efficacy in patients who have exhausted other treatment options.
GnRH agonists and antagonists (leuprolide, goserelin, degarelix) remain mainstays of prostate cancer treatment, suppressing testosterone production to starve androgen-dependent tumors. Somatostatin analogs (octreotide, lanreotide) are standard treatments for neuroendocrine tumors. Peptide-based cancer vaccines, which present tumor-specific peptide epitopes to the immune system, are in clinical trials for melanoma, lung cancer, and other malignancies.
Metabolic Disease
Beyond GLP-1 agonists, several other peptide-based metabolic therapies are advancing. Pramlintide (Symlin), a synthetic analog of the peptide hormone amylin, was approved in 2005 as an adjunct to insulin therapy in diabetes. Amylin is co-secreted with insulin from pancreatic beta cells and helps regulate postprandial glucose by slowing gastric emptying, suppressing glucagon, and promoting satiety.
Cagrilintide, Novo Nordisk's long-acting amylin analog, is being combined with semaglutide in the CagriSema program. The combination targets two distinct appetite-regulation pathways, and Phase 3 data have shown weight loss of approximately 22-24%, comparable to tirzepatide.
Setmelanotide (Imcivree), a melanocortin 4 receptor (MC4R) agonist, was approved in 2020 for rare genetic obesity disorders caused by POMC, PCSK1, or LEPR deficiency. It represents a precision-medicine approach to obesity, treating the specific molecular defect responsible for extreme weight gain in these patients.
Rare Diseases
Peptide drugs have become particularly important in treating rare diseases, where the patient populations are small but the medical need is great. Teduglutide (Gattex), a GLP-2 analog, was approved in 2012 for short bowel syndrome, a condition in which the small intestine is too short to absorb adequate nutrition. Teduglutide promotes intestinal adaptation by stimulating growth of the intestinal mucosa, reducing the need for parenteral nutrition.
Trofinetide (Daybue), a synthetic analog of a naturally occurring tripeptide, became the first FDA-approved treatment for Rett syndrome in 2023. Rett syndrome is a severe neurodevelopmental disorder affecting primarily girls, and the approval of trofinetide was a major milestone for the Rett syndrome community and for peptide therapeutics in neurological disease.
Vosoritide (Voxzogo), a C-type natriuretic peptide analog, was approved in 2021 for achondroplasia, the most common form of dwarfism. It works by stimulating bone growth through the natriuretic peptide receptor, representing a novel mechanism for treating a skeletal disorder.
Infectious Disease
Enfuvirtide (Fuzeon), a 36-amino-acid synthetic peptide, was approved in 2003 for HIV infection. It works by blocking the fusion of HIV with host cells, representing the first entry inhibitor and the first injectable peptide drug for HIV. While its use has declined with the development of more convenient oral antiretrovirals, enfuvirtide remains important for treatment-experienced patients with multi-drug resistant HIV.
Antimicrobial peptides (AMPs) represent a promising frontier for combating antibiotic resistance. The human body produces many natural AMPs (like defensins, cathelicidins, and LL-37) as part of the innate immune response. Synthetic versions of these peptides are being developed as alternatives to conventional antibiotics, with the advantage that bacteria are less likely to develop resistance to peptides that attack their cell membranes through multiple mechanisms simultaneously.
Cardiovascular and Renal Disease
Nesiritide (Natrecor), a recombinant form of human B-type natriuretic peptide (BNP), was approved in 2001 for acute decompensated heart failure. While its clinical use has been debated, it demonstrated the principle that natriuretic peptides could be used therapeutically.
More recently, the cardiovascular benefits of GLP-1 agonists have expanded the role of peptides in cardiology. The SELECT trial's demonstration that semaglutide reduces cardiovascular events in people with obesity (even without diabetes) suggests that peptide-based approaches to cardiovascular risk reduction will grow significantly in the coming years. The FLOW trial's demonstration of kidney protection adds nephrology to the list of specialties where GLP-1 peptides are becoming standard of care.
Dermatology and Cosmetics
Peptides have found widespread application in dermatology and cosmetic science. GHK-Cu (glycyl-L-histidyl-L-lysine copper complex), a naturally occurring tripeptide-copper complex, has been extensively studied for its wound-healing, anti-inflammatory, and skin-remodeling properties. It stimulates collagen synthesis, promotes angiogenesis, and attracts immune cells to wound sites.
Matrixyl (palmitoyl pentapeptide-4), Argireline (acetyl hexapeptide-3), and SNAP-8 (acetyl octapeptide-3) are synthetic peptides widely used in anti-aging skincare. These peptides work through various mechanisms: stimulating collagen production, inhibiting neurotransmitter release at the neuromuscular junction (mimicking a mild botulinum toxin effect), or promoting extracellular matrix remodeling.
Bremelanotide (Vyleesi), a melanocortin receptor agonist, was approved in 2019 for hypoactive sexual desire disorder in premenopausal women. While its primary indication is sexual health, the melanocortin system also plays roles in pigmentation, inflammation, and energy metabolism, suggesting broader applications for melanocortin peptides.
The Research Peptide Landscape
Beyond FDA-approved drugs, a vibrant research peptide landscape includes compounds under active investigation for various health applications. BPC-157 (Body Protection Compound-157), a synthetic pentadecapeptide derived from gastric juice protein sequences, has shown tissue-protective and healing-promoting effects in numerous preclinical studies. Research suggests it may accelerate tendon, ligament, muscle, and bone healing, though large clinical trials are still needed.
Epithalon (epitalon), a synthetic tetrapeptide (Ala-Glu-Asp-Gly) based on the natural peptide epithalamin, is under investigation for potential effects on telomerase activation. Telomerase maintains telomere length, which shortens with each cell division and is associated with cellular aging. Preclinical studies by Vladimir Khavinson and colleagues have suggested that epithalon may stimulate telomerase activity and extend lifespan in animal models.
Thymosin alpha-1 (Zadaxin), a 28-amino-acid peptide originally isolated from thymic tissue, has been approved in over 30 countries for the treatment of hepatitis B and C and as an immune-enhancing agent. It acts by stimulating T-cell maturation, natural killer cell activity, and dendritic cell function. Research is ongoing into its potential applications in cancer immunotherapy and as an adjuvant to vaccines.
CJC-1295 and Ipamorelin are growth hormone-releasing peptides that stimulate the pituitary to produce and release growth hormone through different mechanisms. CJC-1295 is a modified GHRH analog with an extended half-life (achieved through Drug Affinity Complex technology, which enables albumin binding), while Ipamorelin is a selective growth hormone secretagogue that activates the ghrelin receptor. The combination is studied for its potential to enhance growth hormone secretion without significantly increasing cortisol or prolactin levels.
The Market Opportunity
The global peptide therapeutics market has grown dramatically, driven primarily by GLP-1 agonists but also by advances across the full spectrum of peptide-based medicines. Key market figures include:
- Total peptide therapeutics market: approximately $38 billion in 2023, projected to exceed $106 billion by 2033
- GLP-1 agonist market alone: over $40 billion in 2024 (Novo Nordisk + Eli Lilly combined revenues for semaglutide and tirzepatide products)
- Insulin market: approximately $22 billion annually
- Growth hormone market: approximately $4 billion annually
- Over 150 peptide drug candidates in clinical development as of 2024
- Compound annual growth rate (CAGR) of approximately 10.8% projected through 2033
The growth is not limited to established drug companies. A wave of biotech startups focused on peptide therapeutics has attracted billions of dollars in venture capital funding, driven by the GLP-1 success story and by advances in computational design, delivery technology, and manufacturing methods. Companies like Structure Therapeutics, Viking Therapeutics, Terns Pharmaceuticals, and many others are developing next-generation peptide and peptide-like drugs across multiple therapeutic areas.
From Niche to Mainstream
For most of their history, peptide drugs were niche products, prescribed primarily by endocrinologists and oncologists for conditions like diabetes, growth hormone deficiency, and prostate cancer. The GLP-1 era has changed that perception entirely. Semaglutide and tirzepatide are now among the most prescribed drugs in the world, familiar to primary care physicians, cardiologists, and the general public alike. This mainstreaming of peptide therapeutics has created a feedback loop: greater commercial success attracts more investment in peptide drug development, which generates more approved products, which further expands the market.
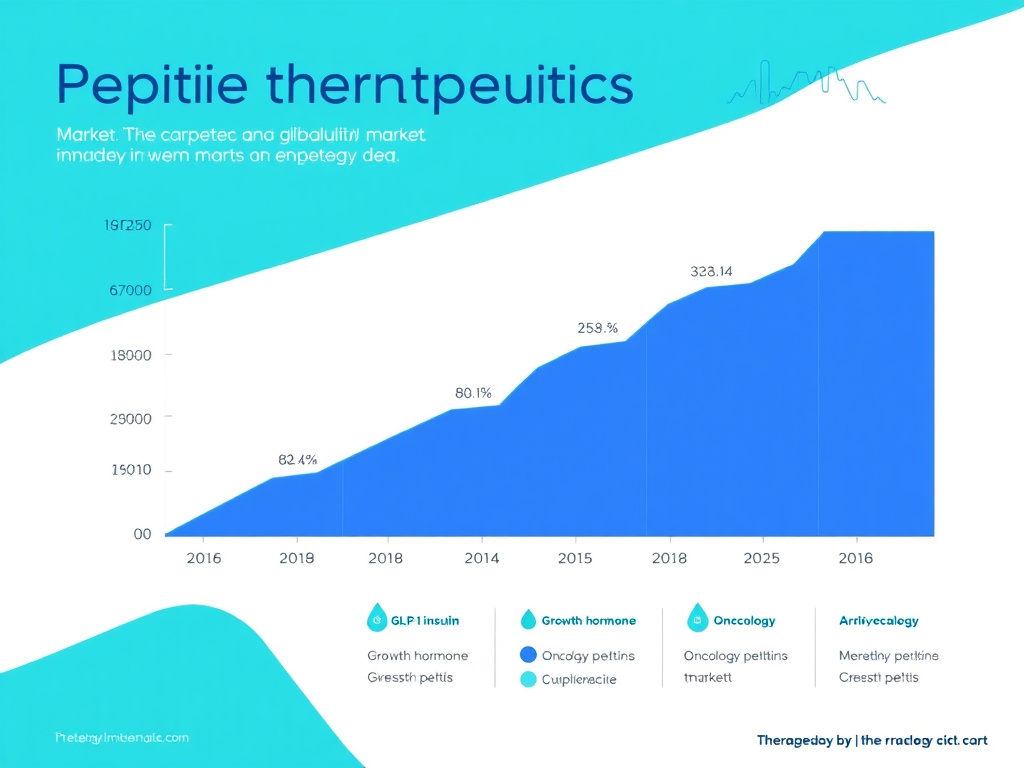
Future Outlook
Peptide medicine is entering its most dynamic period since the discovery of insulin. Multiple converging trends - advances in drug delivery, computational protein design, AI-driven discovery, and expanding clinical indications - suggest that the next decade will produce more peptide drug approvals than any previous decade in history.
Beyond Weight Loss: Expanding GLP-1 Indications
The clinical potential of GLP-1 receptor agonists extends far beyond diabetes and obesity. As of 2024, active clinical trials are investigating semaglutide and related compounds for:
- Non-alcoholic steatohepatitis (NASH/MASH): Semaglutide has shown significant reductions in liver fat and fibrosis scores in Phase 2 trials. Phase 3 trials are ongoing. Survodutide and other dual GLP-1/glucagon agonists are also being tested for this indication.
- Heart failure with preserved ejection fraction (HFpEF): The STEP-HFpEF trial showed that semaglutide improved heart failure symptoms, physical limitations, and exercise function in patients with obesity-related HFpEF.
- Chronic kidney disease: The FLOW trial demonstrated that semaglutide reduced the risk of kidney disease progression by 24% in patients with type 2 diabetes and CKD.
- Obstructive sleep apnea: Tirzepatide significantly reduced the severity of obstructive sleep apnea in the SURMOUNT-OSA trial.
- Addiction: Preclinical and early clinical data suggest GLP-1 agonists may reduce cravings for alcohol, nicotine, and opioids. The mechanism appears to involve modulation of reward pathways in the brain.
- Alzheimer's disease and neurodegeneration: Epidemiological data suggest that GLP-1 agonist users have lower rates of dementia. Clinical trials are testing semaglutide and liraglutide in Alzheimer's disease and Parkinson's disease.
If even a fraction of these indications pan out, the addressable patient population for GLP-1 therapies could expand from hundreds of millions to potentially over a billion people worldwide.
Multi-Agonism: Triple and Beyond
The success of tirzepatide (dual GLP-1/GIP agonist) has validated the multi-agonist approach. Retatrutide, a triple agonist hitting GLP-1, GIP, and glucagon receptors simultaneously, showed even greater weight loss in Phase 2 (up to 24.2% at 48 weeks). Researchers are now exploring whether four-receptor or even broader multi-agonist peptides might further improve efficacy.
The glucagon component in triple agonists is particularly interesting. Glucagon increases energy expenditure by activating thermogenesis in brown adipose tissue and promotes lipid oxidation in the liver. This may explain why retatrutide produces greater weight loss than dual agonists: it adds an energy-burning component on top of the appetite-suppressing effects of GLP-1 and GIP agonism.
Oral Peptide Delivery: Breaking the Injection Barrier
One of the biggest barriers to peptide therapy adoption has been the requirement for injection. Oral semaglutide (Rybelsus) cracked this problem using the SNAC absorption enhancer, but the formulation has limitations: it must be taken on an empty stomach, with restricted water intake, and bioavailability remains low (about 1%).
Next-generation oral delivery approaches aim to overcome these constraints:
- Non-peptide oral GLP-1 agonists: Orforglipron (Eli Lilly) and danuglipron (Pfizer) are small molecules that activate the GLP-1 receptor. They can be taken with food and don't require absorption enhancers. Phase 3 results for orforglipron are expected in 2025.
- Improved absorption enhancers: Companies are developing more efficient permeation enhancers that could increase oral peptide bioavailability from 1% to 5-10%, reducing the dose needed and relaxing dosing restrictions.
- Nanoparticle formulations: Encapsulating peptides in lipid nanoparticles or polymeric carriers can protect them from gastric degradation and facilitate absorption across the intestinal epithelium.
- Device-mediated oral delivery: Ingestible devices (like the SOMA capsule developed at MIT) can inject peptides directly into the gastric or intestinal wall, achieving bioavailabilities comparable to subcutaneous injection.
AI and Computational Design
The 2024 Nobel Prize in Chemistry recognized AlphaFold (Hassabis and Jumper) and computational protein design (Baker). These tools are already beginning to transform peptide drug discovery:
- Structure prediction: AlphaFold can predict the structure of peptide-receptor complexes, enabling rational design of peptides that bind more tightly or selectively to their targets.
- De novo peptide design: David Baker's Rosetta software suite and its successors can design entirely new peptide sequences that fold into desired structures and bind specified targets. This approach can generate leads for peptide drugs without starting from natural peptide sequences.
- Machine learning for ADME prediction: AI models can predict absorption, distribution, metabolism, and excretion properties of peptide candidates, helping to identify compounds with favorable pharmacokinetic profiles early in development.
- Generative models: Large language models trained on protein sequences can generate novel peptide sequences with specified properties, exploring chemical space far more efficiently than traditional combinatorial approaches.
Peptide-Drug Conjugates and Targeted Delivery
Peptide-drug conjugates (PDCs) represent a growing area of research. Like antibody-drug conjugates (ADCs), PDCs use a targeting moiety (in this case, a peptide) to deliver a cytotoxic payload specifically to tumor cells. PDCs offer several advantages over ADCs: they're smaller, cheaper to manufacture, penetrate tissues more readily, and clear from circulation faster (reducing off-target toxicity).
GLP-1 receptor-targeted drug conjugates are also being explored as a way to deliver additional therapeutic agents specifically to tissues that express GLP-1 receptors (pancreatic beta cells, brain, gut). This approach could enable combination therapies that achieve effects impossible with either component alone.
The Broader Peptide Therapeutic Landscape
Beyond GLP-1, other peptide drug classes continue to advance. BPC-157, a peptide derived from gastric juice proteins, is being studied for tissue repair and healing. Epithalon, a synthetic tetrapeptide based on the natural epithalamin, is under investigation for its potential effects on telomerase activity. Thymosin alpha-1 continues to be studied for immune modulation. GHK-Cu, a naturally occurring copper peptide complex, is being explored for wound healing and anti-aging applications.
The peptide drug market is projected to grow from $38 billion in 2023 to over $106 billion by 2033, driven primarily by GLP-1 agonists but also by advances across the full spectrum of peptide therapeutics. With over 150 peptide drugs currently in clinical development, the pipeline has never been larger or more diverse.
Long-Acting Formulations and Depot Technologies
One of the most active areas of peptide drug development is the engineering of ultra-long-acting formulations. While weekly dosing (as with semaglutide and tirzepatide) has been a major improvement over daily injections, monthly or quarterly dosing would further improve patient convenience and adherence. Several approaches are being pursued to achieve this goal.
Subcutaneous depot formulations use biodegradable polymers, lipid-based matrices, or in-situ forming gels to create a reservoir of peptide at the injection site that releases drug gradually over weeks to months. The technology has been proven with leuprolide depot formulations that provide 1, 3, or 6 months of continuous drug release. Applying similar approaches to GLP-1 agonists could yield monthly or quarterly formulations that maintain therapeutic drug levels without the peaks and troughs associated with bolus injections.
Implantable devices offer even longer duration. The ITCA 650 device, developed by Intarcia Therapeutics, was a subcutaneous osmotic mini-pump about the size of a matchstick that could deliver exenatide continuously for 6-12 months. Although the program encountered regulatory difficulties, the concept demonstrated that months-long peptide delivery from a single implant was technically feasible. Similar implant technologies could potentially be adapted for semaglutide, tirzepatide, or next-generation GLP-1 agonists.
Antibody-mediated half-life extension represents another frontier. Bispecific antibodies that bind both a peptide drug and a long-lived serum protein (like albumin or transferrin) could extend peptide half-lives to weeks or months. Several biotech companies are exploring this approach, though no products have yet reached the market.
The ultimate goal of these engineering efforts is a treatment paradigm in which patients with obesity or diabetes receive a peptide drug intervention as infrequently as a few times per year, rather than weekly or daily. Achieving this would dramatically improve adherence, reduce the burden of chronic disease management, and potentially improve long-term outcomes by ensuring more consistent drug exposure.
Personalized Peptide Medicine
One of the most exciting near-term developments is the personalization of peptide therapy. Not all patients respond equally to GLP-1 agonists. In the STEP 1 trial of semaglutide 2.4 mg, while the average weight loss was 14.9%, individual responses ranged from less than 5% to over 30%. Understanding why some patients are "super-responders" while others have modest results is a major research priority.
Genetic factors likely play a role. Variants in the GLP-1 receptor gene (GLP1R) and related signaling pathways may influence how strongly a patient responds to GLP-1 agonist therapy. Genome-wide association studies (GWAS) are being conducted to identify predictive biomarkers. Gut microbiome composition, baseline metabolic status, degree of insulin resistance, and central nervous system sensitivity to GLP-1 may all contribute to response variability.
In the future, clinicians may use pharmacogenomic testing, metabolic profiling, or AI-based prediction models to identify which GLP-1 agonist (or combination of agonists) is most likely to work for a specific patient. A patient with a particular genetic profile might be directed to semaglutide, while another might benefit more from tirzepatide or a triple agonist. This personalized approach could improve outcomes, reduce costs (by avoiding ineffective prescriptions), and minimize side effects.
Personalization extends beyond GLP-1 agonists. For peptide therapies in general, advances in biomarker identification and companion diagnostics could enable precision matching of patients to peptide drugs across oncology, immunology, and other fields. The convergence of AI, genomics, and peptide pharmacology creates a compelling vision of truly personalized peptide medicine.
Combination Therapy Approaches
The future of peptide medicine will likely involve more combinations. CagriSema (semaglutide + cagrilintide) is already demonstrating the power of combining two peptide drugs that target different pathways. Other combinations under investigation include:
- GLP-1 agonists + SGLT2 inhibitors (combining incretin and renal glucose effects)
- GLP-1 agonists + leptin (addressing leptin resistance that accompanies weight loss)
- Multi-agonist peptides + oral metabolic modulators
- GLP-1 agonists + anti-inflammatory peptides for metabolic-associated liver disease
The concept of "metabolic combination therapy" parallels the development of combination antiretroviral therapy for HIV, where using multiple drugs that target different steps in the viral lifecycle proved far more effective than any single agent. Metabolic disease may respond similarly to multi-target approaches.
Novel Delivery Systems Beyond Injection
While oral delivery garners the most attention, other non-invasive delivery routes are being explored for peptide drugs:
- Transdermal patches: Microneedle patches that painlessly deliver peptides through the skin are in development. These patches contain arrays of tiny dissolving needles (less than 1 mm long) that penetrate the outer skin layer and release their peptide payload. Several groups have demonstrated proof-of-concept for insulin delivery via microneedle patches.
- Intranasal delivery: The nasal mucosa provides a large, well-vascularized surface for peptide absorption. Nasal insulin has been tested for Alzheimer's disease (based on evidence that insulin signaling in the brain is impaired in AD), and nasal peptide delivery systems could avoid first-pass metabolism entirely.
- Pulmonary delivery: Inhaled insulin (Exubera, Afrezza) has been marketed, though with limited commercial success. Pulmonary delivery offers rapid absorption and high bioavailability for peptides, and improved inhaler devices and formulations could make this route more practical.
- Implantable devices: Long-lasting implants that release peptide drugs over months are in development. The ITCA 650 device (a matchstick-sized osmotic pump implanted under the skin) was tested for delivering exenatide continuously for 6-12 months, eliminating the need for any injections. While that particular program faced regulatory delays, the concept remains compelling.
The diversity of delivery approaches reflects the breadth of the peptide therapeutics market and the varied needs of different patient populations. A patient who needs daily insulin may prefer an implant or a smart patch that eliminates the need to remember injections. A patient on weekly GLP-1 therapy may be happy with a prefilled pen. And a patient who prefers no injections at all may wait for an effective oral formulation. The availability of multiple delivery options could significantly expand the number of patients who are willing and able to use peptide-based medicines.
What the Next Decade May Bring
- Oral GLP-1 agonists that don't require empty-stomach dosing (2025-2027)
- Triple and multi-agonist peptides with 25%+ weight loss potential (2026-2028)
- GLP-1 agonists approved for Alzheimer's disease, addiction, and liver disease (2027-2030)
- AI-designed peptide drugs entering clinical trials (2025-2028)
- Peptide-drug conjugates for targeted cancer therapy (2025-2030)
- Long-acting depot formulations enabling monthly or quarterly dosing (2026-2030)
The Science of Peptide Drug Design: From Natural Molecules to Engineered Therapeutics
Turning a naturally occurring peptide into a drug requires solving a series of interconnected problems: stability, bioavailability, receptor selectivity, duration of action, and manufacturing scalability. The strategies that peptide scientists have developed to address these challenges constitute one of the most creative chapters in pharmaceutical chemistry.
Why Native Peptides Make Poor Drugs
Most natural peptides have characteristics that make them challenging to use as medicines directly. They are rapidly degraded by peptidases and proteases in the blood, gut, and tissues. Their half-lives are measured in minutes, not hours or days. They are poorly absorbed from the gastrointestinal tract, making oral administration difficult. Many bind to their target receptors with moderate affinity, requiring high doses. And some activate multiple receptor subtypes, leading to unwanted side effects.
Consider native GLP-1 as an example. It has a plasma half-life of less than 2 minutes due to rapid cleavage by DPP-4. Its bioavailability after oral administration is essentially zero. And while it activates the GLP-1 receptor potently, it also stimulates other proglucagon-derived peptide receptors at high concentrations. To turn GLP-1 into a useful drug, researchers had to address all of these limitations.
The same challenges apply, with variations, to virtually every natural peptide that has been considered as a therapeutic candidate. The strategies developed to overcome them can be grouped into several categories.
Strategy 1: Amino Acid Substitution
The simplest approach to improving a peptide's pharmacological properties is to change specific amino acids in its sequence. This can be done to enhance receptor binding, resist protease cleavage, improve solubility, or alter selectivity.
In semaglutide, the replacement of alanine at position 8 with alpha-aminobutyric acid (Aib) was the key modification that conferred DPP-4 resistance. Aib is an alpha-alpha-disubstituted amino acid that creates a steric barrier around the DPP-4 cleavage site. The enzyme simply can't access the bond it needs to cut. This single substitution extended the metabolic stability of semaglutide enormously.
D-amino acid substitutions are another common strategy. Natural proteins use exclusively L-amino acids. Replacing an L-amino acid at a protease-sensitive position with its D-enantiomer (mirror image) often blocks cleavage by proteases, which have evolved to recognize L-amino acid substrates. D-amino acid substitutions must be placed carefully, though, because they can also disrupt the peptide's ability to bind its target receptor if placed at positions critical for receptor interaction.
Non-natural amino acids go further. Hundreds of unnatural amino acids have been incorporated into synthetic peptides, including beta-amino acids (which insert an extra carbon into the backbone), N-methylated amino acids (which block hydrogen bonding at specific positions), and amino acids with modified side chains that confer new properties like fluorescence, click chemistry reactivity, or enhanced hydrophobicity.
Strategy 2: Lipidation and Albumin Binding
One of the most successful strategies for extending peptide half-life is lipidation: attaching fatty acid chains to the peptide. The fatty acid chain binds reversibly to serum albumin, the most abundant protein in blood (concentration about 35-50 g/L, half-life about 19 days). When the lipidated peptide is bound to albumin, it's too large for renal filtration (kidneys filter molecules smaller than about 60 kDa), and it's partially shielded from protease digestion.
Liraglutide uses a C16 palmitic acid attached via a gamma-glutamic acid spacer. This gives it a half-life of about 13 hours, sufficient for once-daily dosing. Semaglutide uses a C18 octadecandioic acid (fatty diacid) attached via a longer linker containing two mini-PEG units and a gamma-glutamic acid. The longer fatty acid and optimized linker provide tighter albumin binding and a half-life of approximately 165 hours (about 7 days), enabling once-weekly dosing.
The albumin-binding approach is elegant because it exploits the body's own protein recycling system. Albumin is continuously internalized by cells through the neonatal Fc receptor (FcRn), recycled in endosomes, and returned to the bloodstream. The lipidated peptide hitchhikes on this recycling pathway, gaining an extended half-life without requiring any chemical modification of the albumin itself.
Insulin detemir (Levemir) and insulin degludec (Tresiba) also use fatty acid acylation to achieve long-acting insulin formulations. Detemir has a 14-carbon myristic acid chain (half-life ~6 hours), while degludec uses a hexadecanedioic acid chain with a gamma-glutamic acid linker (half-life ~25 hours). The same principle, different specific implementations.
Strategy 3: PEGylation
PEGylation involves attaching polyethylene glycol (PEG) chains to a peptide. PEG is a non-toxic, non-immunogenic polymer that increases the effective size of the peptide, reducing renal clearance and shielding it from proteases. PEGylation also improves aqueous solubility of hydrophobic peptides.
PEGylated interferon (Pegasys, PegIntron) and PEGylated growth hormone (Sogroya) are examples of PEGylated peptide/protein therapeutics. The PEG chain can range from a few kDa to 40 kDa or more, with larger PEG chains generally providing longer half-lives but potentially reducing receptor binding affinity due to steric interference.
One limitation of PEGylation is that PEG is not biodegradable. It accumulates in tissues with repeated dosing, and there have been reports of PEG-associated vacuolation in animal studies. This has motivated research into alternatives like polysarcosine, polypeptide-based polymers, and cleavable PEG conjugates that shed their PEG chains after fulfilling their half-life extension function.
Strategy 4: Cyclization
Cyclic peptides, in which the backbone forms a ring rather than a linear chain, are generally more resistant to proteases than their linear counterparts. The ring structure constrains the peptide in a conformation that many proteases can't accommodate in their active sites. Cyclization can also improve membrane permeability by reducing the number of exposed hydrogen bond donors, and it often enhances receptor binding by pre-organizing the peptide in its bioactive conformation.
Nature provides many examples of cyclic peptides with potent biological activity. Cyclosporin A, an 11-residue cyclic peptide from a fungus, is one of the most important immunosuppressive drugs ever developed. Daptomycin (Cubicin), a cyclic lipopeptide antibiotic, is used for serious Gram-positive infections. Octreotide, the somatostatin analog, contains an internal disulfide bond that creates a partial ring structure.
Synthetic chemists have developed numerous strategies for cyclizing peptides: head-to-tail cyclization (connecting the N-terminus to the C-terminus), side-chain-to-side-chain cyclization (using disulfide bonds, lactam bridges, or click chemistry), and stapling (using hydrocarbon bridges across one face of an alpha-helical peptide). Each approach has advantages for specific applications.
Strategy 5: Protein Fusion
Another approach to extending peptide half-life is to fuse the peptide to a larger protein partner. The most common fusion partners are:
- Fc fragment of IgG: The Fc portion of an antibody binds to the neonatal Fc receptor (FcRn), which recycles IgG antibodies and extends their half-life to about 21 days. Dulaglutide (Trulicity) is a GLP-1 analog fused to an IgG4 Fc fragment, giving it a half-life of about 5 days and enabling once-weekly dosing.
- Albumin: Direct genetic fusion of a peptide to human serum albumin gives it the same long half-life as albumin itself. Albiglutide (Tanzeum) was a GLP-1-albumin fusion protein, though it was withdrawn from the market in 2018 due to declining sales rather than safety concerns.
- XTEN: An unstructured, non-immunogenic polypeptide that behaves like a biological PEG. XTEN fusion increases the hydrodynamic radius of the conjugate, reducing renal clearance and extending half-life. Several XTEN-conjugated peptide drugs are in clinical development.
Strategy 6: Controlled-Release Formulations
Physical encapsulation and depot formulations provide another route to extended duration of action. Exenatide extended-release (Bydureon) encapsulates exenatide within biodegradable poly(lactic-co-glycolic acid) (PLGA) microspheres. After subcutaneous injection, the microspheres slowly degrade over weeks, releasing exenatide at a relatively constant rate. This converts a twice-daily injection into a once-weekly injection.
Leuprolide (Lupron Depot) uses a similar microsphere approach to achieve 1-month, 3-month, or even 6-month duration of action from a single injection. This has been highly convenient for patients requiring long-term GnRH agonist therapy for prostate cancer or endometriosis.
More advanced depot technologies are in development, including in-situ forming gels (which are injected as liquids and solidify under the skin), implantable devices (like the histrelin implant for prostate cancer, which lasts 12 months), and bioresorbable matrices that release peptides over periods of months.
Putting It All Together: Semaglutide as a Design Case Study
Semaglutide illustrates how multiple peptide engineering strategies can be combined in a single molecule. Starting from the native GLP-1(7-37) sequence:
- Position 8: Ala replaced with Aib for DPP-4 resistance (amino acid substitution)
- Position 34: Lys replaced with Arg to prevent incorrect acylation (amino acid substitution)
- Position 26: C18 fatty diacid attached via a linker for albumin binding (lipidation)
The result is a molecule with a 165-hour half-life (vs. less than 2 minutes for native GLP-1), potent receptor activation (similar to native GLP-1), and sufficient metabolic stability for once-weekly dosing. Each modification was chosen based on decades of accumulated knowledge about peptide pharmacology. The design process involved synthesizing and testing hundreds of analogs before arriving at the final clinical candidate.
This iterative, structure-driven approach to peptide drug design is the culmination of a century of learning. From Banting and Best's crude pancreatic extracts to Merrifield's automated synthesis to the computationally optimized, multi-modified analogs of today, the trajectory has been consistent: deeper understanding of peptide structure and function enables more precisely engineered therapeutic molecules.
The Half-Life Engineering Challenge
Extending a peptide's half-life from 2 minutes to 7 days, as Novo Nordisk did with semaglutide, required a ~5,000-fold increase in plasma persistence. This was achieved through a combination of DPP-4 resistance (preventing enzymatic degradation), albumin binding (preventing renal filtration and providing a circulating depot), and structural modifications that reduce susceptibility to other proteases. The engineering required to achieve this result was as challenging as any problem in pharmaceutical chemistry, and the clinical impact has been proportionate to the technical difficulty.
Global Impact and Public Health Implications
The century-long history of peptide medicine has profoundly shaped global public health. From saving the lives of millions of people with diabetes to the current potential for addressing the worldwide obesity epidemic, peptide drugs have had an outsized impact on human health relative to other drug classes.
The Diabetes Pandemic and Peptide Solutions
The International Diabetes Federation estimates that 537 million adults (aged 20-79) were living with diabetes in 2021, a number projected to rise to 783 million by 2045. Approximately 90-95% of cases are type 2 diabetes, and the remainder are type 1 and other forms. Diabetes is the ninth leading cause of death globally, directly causing an estimated 6.7 million deaths per year.
Insulin, the original peptide drug, remains essential for all patients with type 1 diabetes (approximately 8.5 million worldwide) and for many with type 2 diabetes whose disease has progressed to the point of beta-cell failure. The WHO added insulin to its Model List of Essential Medicines in 1977, recognizing it as one of the most important drugs in global health.
However, access to insulin remains uneven. In many low- and middle-income countries, insulin is unaffordable or unavailable. The WHO estimates that about half of the people who need insulin for type 2 diabetes don't receive it. The introduction of biosimilar insulins (which have lower manufacturing costs than branded products) and initiatives like the WHO Prequalification Programme for insulin are helping to expand access, but significant gaps remain.
GLP-1 receptor agonists present a different access challenge. While they offer superior efficacy for type 2 diabetes and obesity compared to older therapies, their cost is substantially higher. Semaglutide (Ozempic, Wegovy) and tirzepatide (Mounjaro, Zepbound) can cost $800-$1,300 per month in the United States without insurance, placing them beyond the reach of many patients. The tension between clinical benefit and affordability is one of the defining debates in current healthcare policy.
The Obesity Crisis: Can Peptide Drugs Change the Curve?
Obesity affects approximately 890 million adults worldwide (as of 2022 WHO data), with prevalence rising in virtually every country. Obesity is a major risk factor for type 2 diabetes, cardiovascular disease, certain cancers, sleep apnea, osteoarthritis, and many other conditions. The global economic burden of obesity exceeds $2 trillion per year in direct healthcare costs and lost productivity.
Until the GLP-1 era, pharmacological treatments for obesity were largely ineffective, producing average weight losses of 3-7% of body weight. These modest results, combined with side effects and safety concerns (the fen-phen disaster, sibutramine withdrawal, and others), led many physicians and patients to view anti-obesity drugs with skepticism. Bariatric surgery was the only intervention that reliably produced large, sustained weight loss, but it's invasive, expensive, and not suitable for all patients.
Semaglutide and tirzepatide have fundamentally changed this calculus. Weight losses of 15-22% approach the results of some bariatric surgical procedures (gastric band: 15-25%; sleeve gastrectomy: 20-30%; gastric bypass: 25-35%). What matters even more is that these weight losses translate into meaningful improvements in obesity-related comorbidities: reduced blood pressure, improved lipid profiles, remission of type 2 diabetes, and, as the SELECT trial demonstrated, fewer cardiovascular events.
The question is whether these drugs can be deployed at a scale large enough to meaningfully impact the global obesity epidemic. At current prices and with current supply constraints, the answer is clearly no. But several factors could change this picture:
- Generic and biosimilar competition: Semaglutide's core patents will begin to expire in the late 2020s and early 2030s, potentially opening the door to lower-cost versions.
- Oral formulations: Oral GLP-1 agonists (orforglipron and others) are expected to be cheaper to manufacture than injectable peptides and could significantly expand access.
- Insurance coverage expansion: As cardiovascular and other benefits are demonstrated, payers may become more willing to cover GLP-1 agonists for broader patient populations.
- Public health policy: Some countries may follow the example of the UK and others that have begun integrating GLP-1 agonists into national obesity treatment programs.
Regulatory Evolution: How Drug Agencies Have Adapted to Peptide Therapeutics
The regulatory landscape for peptide drugs has evolved substantially over the past century. When insulin was first used in 1922, no formal drug approval process existed. The FDA, established in 1906, began requiring premarket approval of new drugs only after the Federal Food, Drug, and Cosmetic Act of 1938. Insulin was "grandfathered" in without formal approval.
The 1962 Kefauver-Harris Amendment required manufacturers to demonstrate both safety and efficacy through controlled clinical trials. This framework governed the approval of most peptide drugs through the 1990s. The Biologics Price Competition and Innovation Act of 2009 (BPCIA) created an abbreviated pathway for biosimilar versions of biologic drugs, including peptides. The first biosimilar insulin (Basaglar, biosimilar to Lantus) was approved in 2015.
More recently, the FDA has grappled with issues specific to the GLP-1 class: compounded semaglutide during drug shortages, off-label prescribing of diabetes drugs for weight loss, and the regulatory status of peptides produced by compounding pharmacies versus pharmaceutical manufacturers. These issues reflect the unprecedented demand for GLP-1 agonists and the tension between ensuring drug quality and expanding patient access.
Ethical Considerations
The success of GLP-1 agonists has raised several ethical questions that will shape the future of peptide medicine:
- Equity of access: Should effective anti-obesity drugs be available only to those who can afford them, or does their public health potential justify broader access through insurance coverage or public programs?
- Medicalization of weight: Is treating obesity with lifelong medication appropriate, or should the emphasis be on lifestyle modification and addressing root causes like food systems and built environments?
- Cosmetic versus medical use: How should healthcare systems handle requests for GLP-1 agonists from people who are overweight but not obese? Where is the line between treating a medical condition and enhancing appearance?
- Drug shortages: When demand exceeds supply (as has happened repeatedly with semaglutide and tirzepatide), how should allocation decisions be made? Should patients with diabetes take priority over those seeking weight loss?
- Long-term unknowns: GLP-1 agonists at obesity doses have been studied for only a few years. What are the risks and benefits of lifelong use? What happens when patients stop taking them?
These questions don't have easy answers, but they're central to the responsible development and deployment of peptide therapeutics going forward. The peptide research community and healthcare systems worldwide will need to address them as GLP-1 agonists and other peptide drugs continue to expand in scope and scale.
The Manufacturing Scale Challenge
The explosive demand for GLP-1 agonists has created manufacturing challenges unlike any previously seen in the peptide industry. Novo Nordisk invested over $6 billion in manufacturing expansion between 2022 and 2024, including new production facilities in Denmark, France, and the United States. Eli Lilly similarly committed billions to scaling up tirzepatide production. These investments reflect the sheer volume of peptide that must be produced: with millions of patients on weekly injections of semaglutide or tirzepatide, the annual manufacturing requirement runs into the tons.
Peptide manufacturing at this scale pushes the limits of existing technology. The active pharmaceutical ingredient (API) for semaglutide is produced through a multi-step process involving solid-phase peptide synthesis (or fragment condensation), side-chain acylation with the fatty diacid linker, chromatographic purification, and formulation into the final injectable product. Each step must be performed under GMP (Good Manufacturing Practice) conditions, with rigorous quality control testing at every stage.
The chromatographic purification step is often the bottleneck. Reverse-phase HPLC columns large enough for industrial-scale peptide purification are expensive and have limited throughput. Alternative purification approaches, including multi-column continuous chromatography and membrane-based separations, are being developed to increase capacity. Some manufacturers are also exploring recombinant production of the peptide backbone (expressed in E. coli or yeast) followed by chemical modification, which could be more scalable than fully synthetic approaches for certain peptides.
The supply constraints that have plagued semaglutide and tirzepatide since 2022 highlight a broader issue for the peptide therapeutics industry: as demand grows, manufacturing capacity must grow with it. The capital requirements are enormous, lead times for new facilities are 3-5 years, and the regulatory requirements for pharmaceutical manufacturing are stringent. Companies that have invested early and aggressively in manufacturing capacity are best positioned to capture market share as the GLP-1 class continues to expand.
Environmental Considerations
The environmental footprint of large-scale peptide manufacturing is receiving increasing attention. Traditional SPPS uses large volumes of organic solvents (DMF, DCM, NMP), many of which are classified as substances of very high concern (SVHCs) under European REACH regulations. A typical SPPS synthesis can consume 5,000-15,000 liters of solvent per kilogram of crude peptide produced.
Industry and academic groups are working on greener alternatives. Water-based coupling reactions, solvent recycling and recovery systems, enzymatic ligation methods, and flow chemistry approaches all promise to reduce the environmental impact of peptide manufacturing. Some companies have achieved solvent recovery rates above 90%, significantly reducing waste. The development of more efficient coupling reagents that produce fewer by-products is another active area of research.
The broader question of whether the environmental costs of mass-producing GLP-1 agonists are justified by the health benefits is complex. Obesity and diabetes themselves have enormous environmental implications, from the healthcare system's carbon footprint (hospitals, medical devices, transport) to the agricultural and food processing systems that contribute to metabolic disease. If GLP-1 agonists significantly reduce the prevalence of obesity-related illness, the net environmental impact could actually be positive, but this remains to be rigorously assessed.
Economic Impact: A Multi-Billion Dollar Transformation
The economic transformation driven by GLP-1 agonists extends far beyond the pharmaceutical industry. Key economic effects include:
- Pharmaceutical revenues: Novo Nordisk's semaglutide products generated approximately $21 billion in revenue in 2023, and Eli Lilly's tirzepatide products are on track for similarly massive sales. These revenues have made both companies among the most valuable in the world.
- Healthcare cost offsets: Preliminary analyses suggest that GLP-1 agonist-mediated weight loss and cardiovascular risk reduction could reduce downstream healthcare costs for diabetes complications, cardiovascular events, and obesity-related conditions. However, these cost offsets must be weighed against the high upfront cost of the drugs themselves.
- Food industry disruption: Analysts have begun to quantify the impact of widespread GLP-1 agonist use on food consumption. Some estimates suggest that millions of patients on these drugs consume 20-30% fewer calories, which could have measurable effects on the food, beverage, and restaurant industries.
- Insurance and benefits: Employers and health insurers are grappling with the cost of covering GLP-1 agonists for their enrollees. Some large employers have added coverage; others have restricted it. The total pharmacy spend on GLP-1 agonists in the US is projected to exceed $100 billion annually by the late 2020s if coverage continues to expand.
- Stock market effects: The success of GLP-1 agonists has driven substantial shifts in market capitalization. Novo Nordisk briefly became the most valuable company in Europe. Companies across the food, medical device (bariatric surgery), and adjacent healthcare sectors have seen their valuations affected by the GLP-1 phenomenon.
The Patient Perspective
Behind the scientific achievements and market numbers are the experiences of individual patients whose lives have been changed by peptide medicine. Millions of people with type 1 diabetes owe their survival to insulin, the original peptide drug. Children with growth hormone deficiency have reached normal height thanks to recombinant growth hormone. Patients with prostate cancer have had their disease controlled by GnRH analogs. And now, millions more are experiencing significant weight loss and improved metabolic health through GLP-1 receptor agonists.
Patient surveys consistently show high satisfaction rates with GLP-1 agonists. The most common positive reports include reduced appetite and food noise (the constant preoccupation with food that many people with obesity describe), meaningful weight loss that improves physical function and quality of life, improved blood sugar control, and the psychological relief of finally finding a treatment that works after years of failed diets and exercise programs.
The most common complaints involve gastrointestinal side effects (nausea, vomiting, diarrhea, constipation), which are most pronounced during dose titration and usually improve over time. Cost and insurance coverage are also major concerns, with many patients reporting difficulty affording their medications or facing coverage denials from their insurers.
The patient experience underscores a fundamental truth about peptide medicine: the science matters only if it translates into better outcomes for real people. The century-long arc from Banting's dog experiments in 1921 to the GLP-1 era of the 2020s is, at its core, a story about reducing human suffering through the creative application of peptide chemistry and biology.
The Compounding Pharmacy Dimension
The surge in demand for GLP-1 agonists has created a significant role for compounding pharmacies. When the FDA designates a drug as being in shortage (as it did for semaglutide in 2022-2023 and tirzepatide at various points), compounding pharmacies under Section 503A or 503B of the Federal Food, Drug, and Cosmetic Act can produce compounded versions of the drug to help meet patient needs.
Compounded peptides serve an important function in expanding access, particularly for patients who can't afford or can't obtain the branded products. However, compounded drugs are not FDA-approved, and the quality and consistency of compounded products can vary. Patients considering compounded peptides should work with licensed, accredited pharmacies (ideally those accredited by the Pharmacy Compounding Accreditation Board or similar bodies) and should always consult a healthcare provider. The FormBlends platform provides resources for understanding compounded peptide options and connecting with qualified healthcare providers.
Frequently Asked Questions
References & Clinical Sources
- Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J. 1922;12(3):141-146. doi:10.1016/S0140-6736(01)44940-0
- Sanger F. The free amino groups of insulin. Biochem J. 1945;39(5):507-515. doi:10.1042/bj0390507
- Sanger F, Tuppy H. The amino-acid sequence in the phenylalanyl chain of insulin. Biochem J. 1951;49(4):463-481. doi:10.1042/bj0490463
- du Vigneaud V, Ressler C, Swan JM, Roberts CW, Katsoyannis PG, Gordon S. The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J Am Chem Soc. 1953;75(19):4879-4880. doi:10.1021/ja01115a553
- Merrifield RB. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J Am Chem Soc. 1963;85(14):2149-2154. doi:10.1021/ja00897a025
- Yalow RS, Berson SA. Immunoassay of endogenous plasma insulin in man. J Clin Invest. 1960;39(7):1157-1175. doi:10.1172/JCI104130
- Guillemin R. Peptides in the brain: the new endocrinology of the neuron. Science. 1978;202(4366):390-402. doi:10.1126/science.212832
- Schally AV, Arimura A, Baba Y, et al. Isolation and properties of the FSH and LH-releasing hormone. Biochem Biophys Res Commun. 1971;43(2):393-399. doi:10.1016/0006-291X(71)90766-2
- Hughes J, Smith TW, Kosterlitz HW, et al. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975;258(5536):577-580. doi:10.1038/258577a0
- Goeddel DV, Kleid DG, Bolivar F, et al. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc Natl Acad Sci USA. 1979;76(1):106-110. doi:10.1073/pnas.76.1.106
- Agersø H, Jensen LB, Elbrønd B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45(2):195-202. doi:10.1007/s00125-001-0719-z
- Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409-1439. doi:10.1152/physrev.00034.2006
- Drucker DJ, Habener JF, Holst JJ. Discovery, characterization, and clinical development of the glucagon-like peptides. J Clin Invest. 2017;127(12):4217-4227. doi:10.1172/JCI97233
- Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. J Biol Chem. 1992;267(11):7402-7405. doi:10.1016/S0021-9258(18)42531-8
- Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J Biol Chem. 1986;261(25):11880-11889. doi:10.1016/S0021-9258(18)67324-7
- Knudsen LB, Lau J. The discovery and development of liraglutide and semaglutide. Front Endocrinol. 2019;10:155. doi:10.3389/fendo.2019.00155
- Wilding JPH, Batterham RL, Calanna S, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384(11):989-1002. doi:10.1056/NEJMoa2032183
- Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. 2022;387(3):205-216. doi:10.1056/NEJMoa2206038
- Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and cardiovascular outcomes in obesity without diabetes. N Engl J Med. 2023;389(24):2221-2232. doi:10.1056/NEJMoa2307563
- Frías JP, Davies MJ, Rosenstock J, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6):503-515. doi:10.1056/NEJMoa2107519
- Marso SP, Daniels GH, Tanaka K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311-322. doi:10.1056/NEJMoa1603827
- Jastreboff AM, Kaplan LM, Frías JP, et al. Triple-hormone-receptor agonist retatrutide for obesity. N Engl J Med. 2023;389(6):514-526. doi:10.1056/NEJMoa2301972
- Muttenthaler M, King GF, Adams DJ, Alewood PF. Trends in peptide drug discovery. Nat Rev Drug Discov. 2021;20(4):309-325. doi:10.1038/s41573-020-00135-8
- Wang L, Wang N, Zhang W, et al. Therapeutic peptides: current applications and future directions. Signal Transduct Target Ther. 2022;7(1):48. doi:10.1038/s41392-022-00904-4
- Lau JL, Dunn MK. Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg Med Chem. 2018;26(10):2700-2707. doi:10.1016/j.bmc.2017.06.052
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes. 1995;44(9):1126-1131. doi:10.2337/diab.44.9.1126
- Bliss M. The Discovery of Insulin. University of Chicago Press; 1982. ISBN: 978-0226058993.
- Mitchell AR. Bruce Merrifield and solid-phase peptide synthesis: a historical assessment. Biopolymers. 2008;90(3):175-184. doi:10.1002/bip.20925
- Nauck MA, Quast DR, Wefers J, Meier JJ. GLP-1 receptor agonists in the treatment of type 2 diabetes: state-of-the-art. Mol Metab. 2021;46:101102. doi:10.1016/j.molmet.2020.101102
- Rosenstock J, Wysham C, Frías JP, et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1). Lancet. 2021;398(10295):143-155. doi:10.1016/S0140-6736(21)01324-6
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315-1317. doi:10.1126/science.4001944
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583-589. doi:10.1038/s41586-021-03819-2
- Hodgkin DC. The X-ray analysis of the structure of penicillin. Adv Sci. 1949;6(22):85-89.
- Brown JC, Mutt V, Pederson RA. Further purification of a polypeptide demonstrating enterogastrone activity. J Physiol. 1970;209(1):57-64. doi:10.1113/jphysiol.1970.sp009155
- Blundell TL, Cutfield JF, Cutfield SM, et al. Atomic positions in rhombohedral 2-zinc insulin crystals. Nature. 1971;231(5304):506-511. doi:10.1038/231506a0
- Gerber PP, Rutter GA. GLP-1 receptor agonists in cardiovascular disease: current evidence and future directions. Cardiovasc Diabetol. 2023;22(1):314. doi:10.1186/s12933-023-02047-2
- Kosiborod MN, Abildstrøm SZ, Borlaug BA, et al. Semaglutide in patients with heart failure with preserved ejection fraction and obesity. N Engl J Med. 2023;389(12):1069-1084. doi:10.1056/NEJMoa2306963
- Perkovic V, Tuttle KR, Rossing P, et al. Effects of semaglutide on chronic kidney disease in patients with type 2 diabetes. N Engl J Med. 2024;391(2):109-121. doi:10.1056/NEJMoa2403347
- Al Musaimi O, Al Shaer D, de la Torre BG, Albericio F. 2024 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals. 2025;18(3):291. doi:10.3390/ph18030291
- Drucker DJ. GLP-1 physiology informs the pharmacotherapy of obesity. Mol Metab. 2022;57:101351. doi:10.1016/j.molmet.2021.101351
For the complete FormBlends peptide research library, visit our science page or explore specific compounds including semaglutide, tirzepatide, BPC-157, CJC-1295/Ipamorelin, Epithalon, GHK-Cu, and Thymosin Alpha-1.