Executive Summary

Key Takeaways
- First-in-class: SS-31 is the first mitochondria-targeted peptide to achieve FDA approval (September 2025, as FORZINITY for Barth syndrome)
- Mechanism: Selectively binds cardiolipin on the inner mitochondrial membrane, stabilizing electron transport chain complexes and reducing oxidative stress
- Rapid uptake: Concentrates in mitochondria at 1,000-5,000x cytoplasmic levels within minutes of administration
- Broad potential: Under investigation for heart failure, macular degeneration, primary mitochondrial myopathy, renal disease, and age-related decline
- Safety profile: Generally well tolerated across clinical trials, with injection site reactions as the most common adverse event
SS-31 (elamipretide) is a synthetic tetrapeptide that represents the first mitochondria-targeted drug to receive FDA approval. By selectively binding cardiolipin on the inner mitochondrial membrane, SS-31 stabilizes the electron transport chain, reduces oxidative stress, and restores ATP production in dysfunctional mitochondria. Its September 2025 approval under the brand name FORZINITY for Barth syndrome marked a turning point for mitochondrial medicine.
Mitochondrial dysfunction sits at the center of dozens of diseases, from heart failure to neurodegenerative conditions to the basic biology of aging itself. For decades, researchers understood that failing mitochondria drove disease progression, but they lacked tools precise enough to intervene at the organelle level. SS-31 (elamipretide) changed that calculus. Developed from a family of Szeto-Schiller peptides first characterized in the early 2000s, this four-amino-acid compound crosses cell membranes within minutes, concentrates in mitochondria at levels 1,000 to 5,000 times higher than surrounding cytoplasm, and directly interacts with cardiolipin, the signature phospholipid of the inner mitochondrial membrane.
The clinical significance of this mechanism cannot be overstated. Cardiolipin anchors the protein complexes of the electron transport chain. When cardiolipin becomes oxidized or destabilized, whether from genetic mutations as in Barth syndrome, ischemic damage as in heart attacks, or the cumulative wear of aging, mitochondria lose their ability to produce ATP efficiently. They generate excess reactive oxygen species. They swell, fragment, and eventually trigger cell death pathways. SS-31 intervenes at this exact point, preventing cardiolipin peroxidation, maintaining cristae architecture, and preserving the functional integrity of the respiratory chain.
Clinical trials spanning more than a decade have tested elamipretide across a range of conditions. The TAZPOWER trial in Barth syndrome provided the efficacy and safety data that led to FDA accelerated approval. EMBRACE-STEMI explored cardioprotection during acute myocardial infarction. PROGRESS-HF tested the peptide in chronic heart failure with reduced ejection fraction. The ReCLAIM program investigated its potential in dry age-related macular degeneration. And a growing body of preclinical evidence suggests that SS-31 may address fundamental aging biology by restoring mitochondrial function in aged tissues throughout the body.
The story of SS-31 extends beyond any single disease. It represents proof of concept that targeting mitochondria directly can produce measurable clinical benefits. This report examines the peptide's development history, its molecular mechanism, the clinical evidence across multiple indications, and its broader implications for the emerging field of mitochondrial therapeutics. For individuals exploring peptide-based approaches to cellular health, understanding elamipretide's science provides a foundation for appreciating how mitochondrial function shapes overall vitality and disease risk.
Key Takeaways
- First-in-class: SS-31 is the first mitochondria-targeted peptide to achieve FDA approval (September 2025, as FORZINITY for Barth syndrome)
- Mechanism: Selectively binds cardiolipin on the inner mitochondrial membrane, stabilizing electron transport chain complexes and reducing oxidative stress
- Rapid uptake: Concentrates in mitochondria at 1,000-5,000x cytoplasmic levels within minutes of administration
- Broad potential: Under investigation for heart failure, macular degeneration, primary mitochondrial myopathy, renal disease, and age-related decline
- Safety profile: Generally well tolerated across clinical trials, with injection site reactions as the most common adverse event
The approval of elamipretide for Barth syndrome validated the mitochondrial targeting approach, but it also opened the door to a much larger conversation. Mitochondrial dysfunction contributes to cardiovascular disease, neurodegenerative conditions, metabolic syndrome, and the basic biology of aging. If a peptide can rescue mitochondrial function in the rare and severe context of Barth syndrome, the logical question becomes: what can it do in the far more common conditions where mitochondria fail more gradually? That question drives the ongoing research covered throughout this report, and it connects SS-31 to the broader field of peptide therapeutics aimed at restoring cellular function from the inside out.
Szeto-Schiller Peptide Development

Figure 2: The development journey of SS-31 from academic discovery in the early 2000s through FDA approval in 2025, spanning more than two decades of research and clinical testing.
The Szeto-Schiller peptides emerged from a collaboration between Hazel H. Szeto at Weill Cornell Medical College and Peter W. Schiller at the Clinical Research Institute of Montreal, representing one of the most successful examples of rational drug design targeting subcellular organelles.
Origins of the SS Peptide Family
The story of SS-31 begins in the late 1990s and early 2000s, when Hazel Szeto and Peter Schiller set out to design small peptides capable of penetrating cell membranes and concentrating within mitochondria. At the time, conventional wisdom held that targeting drugs to specific organelles inside living cells was an extraordinarily difficult proposition. Most drug molecules either couldn't cross the plasma membrane efficiently, or if they could, they distributed randomly throughout the cell without preferential accumulation in any particular compartment.
Szeto and Schiller took a different approach. They recognized that the inner mitochondrial membrane possessed a unique lipid composition, dominated by the doubly-charged phospholipid cardiolipin, which created a strongly negative electrostatic environment. By designing small peptides with alternating aromatic and basic amino acid residues, they could create molecules that were both cell-permeable (thanks to their small size and lipophilicity) and mitochondria-selective (thanks to electrostatic attraction between positively charged residues and negatively charged cardiolipin).
The result was a family of tetrapeptides designated SS-01 through SS-31, with "SS" standing for Szeto-Schiller. Each variant differed in its specific amino acid sequence, but all shared the core structural motif: alternating aromatic and cationic residues within a four-amino-acid framework. The most promising of these, SS-31, had the sequence D-Arg-2',6'-dimethyltyrosine-Lys-Phe-NH2. The inclusion of D-arginine (a non-natural stereoisomer) at position one conferred resistance to enzymatic degradation, while the dimethyltyrosine at position two provided a potent aromatic residue with antioxidant properties.
Early Preclinical Characterization
The initial publications describing the SS peptide family appeared between 2004 and 2006, and the data were striking. Zhao and colleagues demonstrated that SS-31 accumulated in mitochondria at concentrations thousands of times higher than in the surrounding cytoplasm, regardless of mitochondrial membrane potential. This was a critical distinction from earlier attempts at mitochondrial targeting using lipophilic cations like triphenylphosphonium (TPP+), which required an intact membrane potential for uptake. Because SS-31's mitochondrial targeting depended on cardiolipin binding rather than membrane potential, the peptide could reach mitochondria even in damaged or depolarized cells, exactly the cells where intervention was most needed.
Early mechanistic studies focused on SS-31's antioxidant properties. The dimethyltyrosine residue could scavenge reactive oxygen species directly, and the peptide's localization at the inner mitochondrial membrane placed it at the precise site where most cellular ROS production occurs. But as research progressed, it became clear that simple ROS scavenging accounted for only a fraction of SS-31's biological activity. The peptide's interaction with cardiolipin produced effects that went far beyond antioxidation.
Work by Birk and colleagues showed that SS-31 stabilized the interaction between cardiolipin and cytochrome c, the mobile electron carrier that shuttles electrons between Complex III and Complex IV of the respiratory chain. When cardiolipin binds cytochrome c, it regulates the protein's electron transfer activity. SS-31 optimized this interaction, promoting cytochrome c's role as an electron carrier while inhibiting its peroxidase activity, which would otherwise catalyze cardiolipin oxidation. This dual effect created a positive feedback loop: by protecting cardiolipin from oxidation, SS-31 maintained the conditions necessary for efficient electron transport, which in turn reduced ROS production, which further protected cardiolipin.
Preclinical Disease Models: Building the Case
Between 2006 and 2010, the Szeto laboratory and collaborators tested SS-31 in an expanding array of preclinical disease models. The peptide showed protective effects in models of cardiac ischemia-reperfusion injury, where it reduced infarct size and preserved left ventricular function when administered before or during reperfusion. In models of acute kidney injury, SS-31 attenuated tubular damage and accelerated the recovery of renal function. In neurological models, it protected against neuronal death following focal cerebral ischemia and reduced damage in models of Parkinson's disease.
Perhaps most provocatively, SS-31 showed remarkable effects in aged animals. When administered to old mice, the peptide reversed age-related declines in mitochondrial function within skeletal muscle, improved cardiac diastolic function, and restored redox homeostasis without requiring an increase in mitochondrial biogenesis. These findings suggested that aged mitochondria weren't irreversibly damaged; they were dysfunctional in a way that could be corrected by stabilizing their membrane architecture.
The breadth of these preclinical results reflected a fundamental truth about mitochondrial dysfunction: it wasn't specific to any single organ or disease. It was a common thread running through conditions as different as heart attacks and aging. A drug that could restore mitochondrial function at its source, by stabilizing the lipid environment of the inner membrane, had the potential to be broadly therapeutic. Related peptides with different mechanisms, like Humanin and MOTS-c, have also shown promise in mitochondrial-related conditions, reflecting the growing recognition that mitochondrial function represents a key therapeutic target.
From Academic Lab to Biotech Company
The clinical potential of SS-31 attracted attention from both the academic community and the pharmaceutical industry. In 2006, Hazel Szeto founded Stealth Peptides Inc. (later renamed Stealth BioTherapeutics) to develop SS-31 and related compounds for clinical use. The company licensed the peptide technology from the Cornell Research Foundation and began the process of pharmaceutical development necessary for human trials.
The company adopted the name "elamipretide" as the International Nonproprietary Name (INN) for SS-31, with "Bendavia" serving as an early clinical formulation name. The compound also carried the research designations MTP-131 in some clinical contexts. Despite these multiple names, all refer to the same tetrapeptide: D-Arg-Dmt-Lys-Phe-NH2.
Stealth BioTherapeutics raised significant capital through multiple funding rounds and an eventual IPO on the Nasdaq exchange, reflecting investor confidence in the mitochondrial targeting platform. The company's development strategy was ambitious, pursuing multiple clinical indications simultaneously: heart failure, Barth syndrome, primary mitochondrial myopathy, dry age-related macular degeneration, and renal disease. This broad pipeline reflected the underlying science, which suggested that cardiolipin stabilization could benefit any tissue where mitochondrial dysfunction drove disease pathology.
Entering Clinical Development
SS-31 entered clinical trials in 2010, marking the transition from bench to bedside for the Szeto-Schiller peptide family. Phase 1 studies established the safety and pharmacokinetic profile of the compound in healthy volunteers. The peptide was administered both intravenously (for acute settings like myocardial infarction) and subcutaneously (for chronic conditions requiring daily dosing), with both routes showing acceptable tolerability.
The pharmacokinetic profile revealed several favorable characteristics. After subcutaneous injection, elamipretide reached peak plasma concentrations within approximately one hour. The half-life was relatively short (approximately 2-3 hours in plasma), but the relevant pharmacological effect was determined by the peptide's residence time within mitochondrial membranes rather than its plasma concentration. Studies using radiolabeled SS-31 showed that the peptide remained associated with cardiolipin-rich membranes for extended periods even after plasma levels declined, supporting once-daily dosing for chronic indications.
By 2012, Stealth BioTherapeutics had initiated Phase 2 trials across multiple indications, launching what would become one of the most comprehensive clinical programs in the history of mitochondrial medicine. The peptide research hub covers the broader context of how peptide therapeutics are advancing across various therapeutic areas.
Regulatory Milestones and FDA Approval
The regulatory path for elamipretide was shaped by several key designations. The FDA granted Orphan Drug Designation for Barth syndrome and primary mitochondrial myopathy, recognizing these as rare diseases with unmet medical needs. The FDA also granted Fast Track Designation and Rare Pediatric Disease Designation for the Barth syndrome indication, expediting the review process.
On September 19, 2025, the FDA granted accelerated approval to FORZINITY (elamipretide) injection for the improvement of muscle strength in adult and pediatric patients with Barth syndrome weighing at least 30 kilograms. This made elamipretide the first FDA-approved treatment specifically for Barth syndrome and, more broadly, the first approved mitochondria-targeted therapeutic. The approval was based on the knee extensor muscle strength data from the open-label extension of the TAZPOWER trial, with continued approval contingent upon verification of clinical benefit in confirmatory trials.
The journey from Szeto and Schiller's academic laboratory to FDA approval took over two decades, spanning basic science discovery, extensive preclinical testing, multiple clinical trial programs, and regulatory review. It stands as an example of how fundamental research into cellular biology can eventually yield clinically meaningful therapeutics, even when the path is long and nonlinear.
Key Milestones in SS-31 Development Timeline
| Year | Milestone | Significance |
|---|---|---|
| 2000-2003 | Szeto and Schiller design and synthesize SS peptide family | First cell-permeable mitochondria-targeted peptides created through rational design |
| 2004 | First publications characterizing SS-31 (Zhao et al.) | Demonstrated 1,000-5,000x mitochondrial accumulation independent of membrane potential |
| 2006 | Stealth Peptides Inc. founded by Hazel Szeto | Technology licensed from Cornell Research Foundation for clinical development |
| 2007-2009 | Extensive preclinical testing across disease models | Cardioprotection, renal protection, and neuroprotection demonstrated in animal models |
| 2010 | First-in-human Phase 1 clinical trials begin | Safety and PK profile established for both IV and SC administration |
| 2012-2014 | Phase 2 trials launched across multiple indications | Heart failure, Barth syndrome, mitochondrial myopathy, and AMD programs initiated |
| 2015 | Orphan Drug Designation granted for Barth syndrome | Regulatory pathway accelerated for rare disease indication |
| 2016 | EMBRACE-STEMI results published | Mixed results in acute MI but reduced incidence of heart failure post-PCI |
| 2017 | Early-phase HF trial published showing LV volume improvements | First evidence of acute cardiac effects in heart failure patients |
| 2018 | TAZPOWER enrollment completed | Barth syndrome Phase 2/3 trial fully enrolled |
| 2020 | PROGRESS-HF results published | 4-week treatment did not meet primary LVESV endpoint in HFrEF |
| 2021 | TAZPOWER crossover results published | 12-week blinded phase did not meet primary endpoint; OLE initiated |
| 2023 | MMPOWER-3 results published | Phase 3 PMM trial did not meet primary endpoints |
| 2024 | 168-week TAZPOWER OLE data published | Long-term improvements in 6MWT, muscle strength, cardiac function confirmed |
| September 2025 | FDA accelerated approval of FORZINITY | First mitochondria-targeted therapeutic approved; first Barth syndrome treatment |
The regulatory journey also highlighted the challenges of developing drugs for rare mitochondrial diseases. Patient identification and recruitment required collaboration with the Barth Syndrome Foundation and an international network of mitochondrial disease specialists. The ultra-rare nature of the condition (approximately 150 known patients in the US) meant that conventional large-scale trial designs were impossible, necessitating innovative statistical approaches and long-term open-label extensions to generate sufficient evidence of efficacy.
The financial journey of Stealth BioTherapeutics itself mirrored the scientific one. The company raised substantial venture capital before going public on Nasdaq, but the series of neutral clinical trial results (EMBRACE-STEMI, PROGRESS-HF, MMPOWER-3, ReCLAIM-2) tested investor patience. The eventual FDA approval for Barth syndrome validated the underlying science and the company's persistence, though the commercial opportunity in an ultra-rare disease remains limited compared to the larger indications that had been the original focus of development.
For context on how other peptide therapeutics have navigated the research-to-approval pipeline, tirzepatide and retatrutide represent examples of peptide drugs that have achieved or are pursuing regulatory approval through large-scale cardiovascular and metabolic trials. The Retatrutide Hub covers the development trajectory of that triple agonist peptide.
Mitochondrial Targeting Mechanism
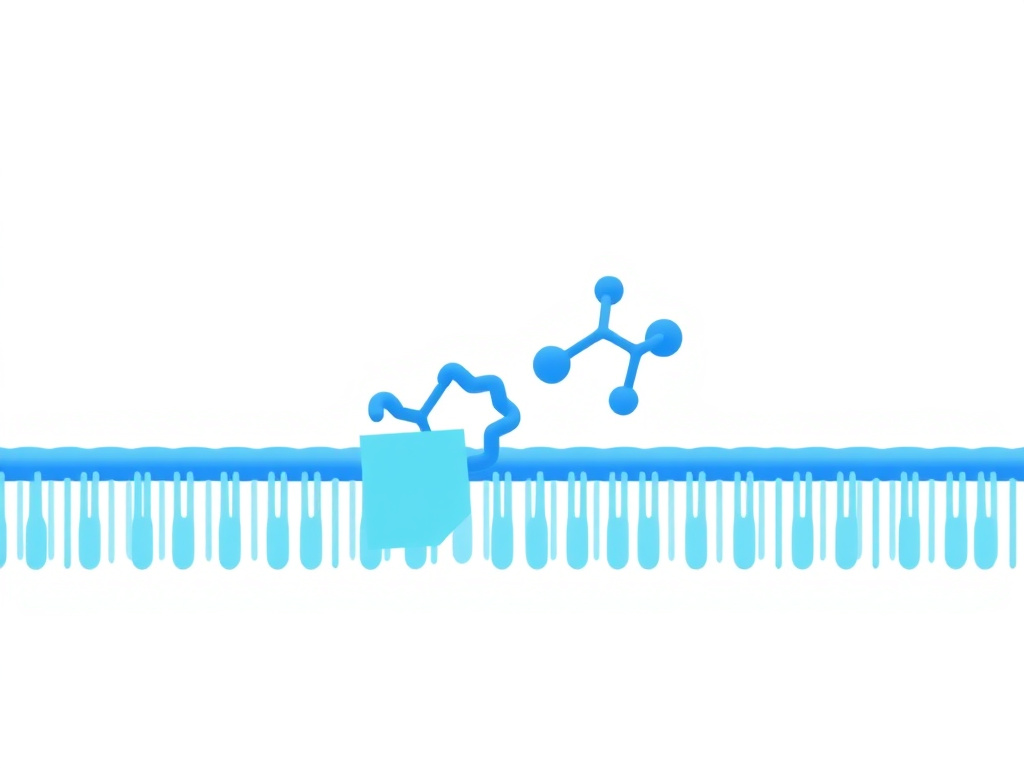
SS-31 achieves its therapeutic effects through a unique cell-penetrating mechanism that concentrates the peptide at the inner mitochondrial membrane, where it interacts directly with cardiolipin to modulate electron transport chain function, reduce ROS production, and restore ATP synthesis.
Cell-Penetrating Properties
Understanding how SS-31 reaches its intracellular target requires appreciating its structural chemistry. The tetrapeptide D-Arg-Dmt-Lys-Phe-NH2 carries a net charge of +3 at physiological pH, with the two basic residues (D-arginine and lysine) each contributing a positive charge, and the amidated C-terminus adding another. Despite this positive charge, which would normally prevent a molecule from crossing the lipid bilayer, SS-31 is highly cell-permeable.
This apparent paradox is resolved by the peptide's alternating aromatic-cationic structural motif. The aromatic residues (dimethyltyrosine and phenylalanine) create hydrophobic faces that can interact with the lipid core of cell membranes, while the cationic residues interact with the negatively charged phospholipid headgroups. This amphipathic character allows SS-31 to partition into and across lipid bilayers through a mechanism that doesn't require active transport or receptor-mediated endocytosis.
Uptake studies using fluorescently labeled SS-31 have demonstrated that the peptide enters cells within minutes of exposure. This rapid uptake occurs across a wide range of cell types, including cardiomyocytes, neurons, renal tubular cells, retinal pigment epithelial cells, and skeletal muscle fibers. The universality of uptake reflects the fundamental physics of membrane partitioning rather than any cell-type-specific transport mechanism.
Mitochondrial Accumulation: Beyond Membrane Potential
Once inside the cell, SS-31 accumulates selectively in mitochondria. This selectivity has been confirmed using multiple techniques: confocal microscopy with fluorescent analogs, subcellular fractionation with mass spectrometric quantification, and live-cell imaging with mitochondrial co-localization markers. The concentration gradient is impressive: SS-31 reaches levels 1,000 to 5,000 times higher within mitochondria than in the surrounding cytoplasm.
What makes this accumulation particularly significant is its independence from mitochondrial membrane potential. Previous strategies for targeting molecules to mitochondria relied on the large negative membrane potential (approximately -180 mV) across the inner mitochondrial membrane, which drives the accumulation of lipophilic cations according to the Nernst equation. Compounds like triphenylphosphonium (TPP+) and Mito-Q exploit this electrical gradient to achieve mitochondrial targeting. But this approach has a fundamental limitation: in diseased or damaged mitochondria, the membrane potential collapses, and potential-dependent targeting fails precisely when it is needed most.
SS-31 avoids this problem entirely. Its mitochondrial accumulation depends not on membrane potential but on its affinity for cardiolipin, which is present exclusively in the inner mitochondrial membrane (and to a lesser extent, at contact sites between the inner and outer membranes). Cardiolipin constitutes approximately 20% of the total lipid content of the inner mitochondrial membrane, creating a dense target for a peptide designed to bind it. Even when mitochondria are depolarized, their cardiolipin content remains, and SS-31 can still reach and stabilize the inner membrane.
This potential-independent targeting mechanism is therapeutically critical. In conditions like ischemia-reperfusion injury, where mitochondrial membrane potential collapses during ischemia and then surges during reperfusion (driving ROS production), SS-31 can be present at the inner membrane throughout the injury cycle. In aging, where mitochondrial membrane potential gradually declines, SS-31 can still access its target. This makes SS-31 fundamentally different from, and complementary to, other mitochondrial-targeting strategies.
The Cardiolipin Binding Interface
The molecular details of how SS-31 interacts with cardiolipin have been elucidated through a combination of biophysical studies, molecular dynamics simulations, and genetic approaches. Cardiolipin is a unique phospholipid with a dimeric structure: two phosphatidic acid molecules linked through a central glycerol backbone, giving it four acyl chains and two negatively charged phosphate groups. In the inner mitochondrial membrane, cardiolipin adopts specific conformations that are essential for its biological functions, including the formation of cristae junctions, the stabilization of respiratory chain supercomplexes, and the regulation of mitochondrial fission and fusion.
SS-31 binds to cardiolipin through a combination of electrostatic and hydrophobic interactions. The positively charged D-arginine and lysine residues interact with the negatively charged phosphate headgroups, while the aromatic dimethyltyrosine and phenylalanine residues insert into the hydrophobic region near the acyl chain tails. This binding mode positions SS-31 at the interface between the aqueous phase and the lipid bilayer, exactly where the headgroups of cardiolipin interact with the peripheral domains of electron transport chain proteins.
Research published in the Journal of Biological Chemistry demonstrated that SS-31 modulates the surface electrostatics of lipid bilayers containing cardiolipin. By altering the charge distribution at the membrane surface, SS-31 influences how peripheral membrane proteins interact with the inner membrane, effectively tuning the activity of the electron transport chain. This electrostatic modulation represents a more sophisticated mechanism than simple antioxidant activity, and it explains why SS-31 can produce effects that stoichiometric ROS scavengers cannot match.
Experimental Evidence for the Binding Mechanism
The molecular details of SS-31's interaction with cardiolipin have been established through multiple complementary experimental approaches. Nuclear magnetic resonance (NMR) spectroscopy studies have characterized the conformational changes in both the peptide and cardiolipin upon binding. Surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) have quantified the binding affinity and thermodynamic parameters. And molecular dynamics (MD) simulations have provided atomistic views of how the peptide positions itself at the lipid-water interface.
NMR studies revealed that SS-31 adopts a defined conformation when bound to cardiolipin-containing membranes, with the aromatic residues (Dmt and Phe) inserting into the acyl chain region while the cationic residues (D-Arg and Lys) remain at the headgroup interface. This positioning places the peptide in what might be called a "sentinel" position: deep enough to interact with the vulnerable acyl chains but exposed enough to interact with peripheral membrane proteins like cytochrome c.
The binding affinity of SS-31 for cardiolipin is in the low micromolar range, which is sufficiently strong for selective accumulation at cardiolipin-rich membranes but not so strong that the peptide becomes permanently stuck. This "Goldilocks" affinity allows dynamic interaction with the membrane, permitting lateral diffusion along the membrane surface and transient interactions with multiple cardiolipin molecules and membrane proteins. This dynamic binding is thought to be important for the peptide's ability to influence multiple cardiolipin-dependent processes simultaneously rather than getting sequestered at a single binding site.
Coarse-grained MD simulations by Mitchell and colleagues (2020) provided insight into how SS-31 modulates the physical properties of cardiolipin-containing bilayers. They showed that the peptide alters the surface charge distribution, changes the lateral pressure profile, and modulates membrane curvature in cardiolipin-enriched domains. These biophysical changes have downstream effects on protein-lipid interactions, helping explain how a small peptide can produce widespread functional changes in the electron transport chain.
Effects on the Electron Transport Chain
The electron transport chain (ETC) consists of four multi-protein complexes (I through IV) plus ATP synthase (Complex V), all embedded in or associated with the inner mitochondrial membrane. Cardiolipin plays essential structural and functional roles in all of these complexes. It stabilizes the quaternary structure of individual complexes, facilitates the formation of supercomplexes (respirasomes), and provides essential co-factor interactions that support electron transfer.
When SS-31 binds to cardiolipin in the inner mitochondrial membrane, it produces several measurable effects on ETC function. First, it improves the coupling efficiency between electron transport and ATP synthesis. In damaged or aged mitochondria, electron leak at Complexes I and III generates superoxide radicals rather than contributing to the proton motive force that drives ATP synthesis. SS-31 reduces this electron leak by stabilizing the structural integrity of these complexes, ensuring that a higher proportion of electron flow contributes to productive ATP generation.
Second, SS-31 promotes the assembly and stability of supercomplexes. Research has shown that the respiratory chain complexes don't function as isolated units; they assemble into higher-order supercomplexes that channel electrons more efficiently and reduce the probability of premature electron escape to molecular oxygen. Cardiolipin is essential for supercomplex formation, and its oxidation disrupts these assemblies. By protecting cardiolipin from oxidation, SS-31 maintains supercomplex integrity and the functional benefits it provides.
Third, SS-31 optimizes the interaction between cardiolipin and cytochrome c, the mobile electron carrier that connects Complex III to Complex IV. Cytochrome c binds cardiolipin on the outer surface of the inner membrane, and this interaction regulates both its electron carrier function and its role as a trigger for apoptosis. Under normal conditions, cardiolipin-bound cytochrome c functions as an efficient electron shuttle. When cardiolipin becomes oxidized (peroxidized), the cytochrome c-cardiolipin interaction shifts: cytochrome c develops peroxidase activity that further oxidizes cardiolipin, creating a vicious cycle that ultimately leads to the release of cytochrome c into the cytoplasm and activation of the apoptotic cascade. SS-31 prevents this shift by protecting cardiolipin from peroxidation, keeping cytochrome c in its electron carrier conformation and preventing the initiation of programmed cell death.
Beyond ROS Scavenging: The Expanded Mechanism
Early descriptions of SS-31's mechanism emphasized its antioxidant properties, particularly the ability of the dimethyltyrosine residue to scavenge reactive oxygen species directly. While this is a real property of the molecule, subsequent research has demonstrated that direct ROS scavenging accounts for only a minor fraction of SS-31's protective effects.
A landmark study published in PNAS by Pharaoh and colleagues (2020) used a comprehensive proteomic approach to map the mitochondrial protein interaction field of SS-31. They found that the peptide interacted with proteins involved in the electron transport chain, the TCA cycle, fatty acid oxidation, amino acid metabolism, and mitochondrial protein import. These interactions were predominantly mediated through cardiolipin-dependent protein-lipid contacts, confirming that SS-31's primary mechanism is modulation of cardiolipin-protein interactions rather than direct chemical antioxidation.
More recently, research has identified additional mechanisms including modulation of the adenine nucleotide translocator (ANT), which exchanges ATP and ADP across the inner mitochondrial membrane. Campbell and colleagues (2023) demonstrated that elamipretide improved ADP sensitivity in aged muscle mitochondria by increasing uptake through the ANT, directly improving the coupling between ATP demand and mitochondrial ATP production. This finding added another dimension to SS-31's mechanism and helped explain its effects on muscle function and exercise tolerance.
For those interested in other compounds that target cellular energy pathways, NAD+ and 5-Amino-1MQ represent complementary approaches to restoring metabolic function, working through different but related biochemical pathways. The Science & Research page provides additional context on the relationship between cellular energetics and health outcomes.
Dose-Response Relationships and Therapeutic Window
Understanding the dose-response relationship of SS-31 is important for both clinical dosing and for appreciating the compound's pharmacological properties. In vitro studies have shown a concentration-dependent improvement in mitochondrial function, with effects beginning at nanomolar concentrations and reaching maximal effect in the low micromolar range. At very high concentrations (above 100 micromolar in some assay systems), SS-31 can actually impair mitochondrial function, likely by disrupting normal cardiolipin organization at the membrane surface through excessive binding. This creates an inverted-U dose-response curve with a well-defined therapeutic window.
In vivo, the therapeutic window is comfortably wide. The approved clinical dose of 40 mg subcutaneous achieves peak plasma concentrations well within the efficacious range, and even at the highest IV doses tested in clinical trials (0.25 mg/kg/hr for 4 hours), there was no evidence of toxicity or impaired mitochondrial function. The rapid clearance of SS-31 from plasma (half-life of 2-3 hours) provides an additional safety margin, as any excess peptide is quickly eliminated even though the therapeutically relevant mitochondrial membrane concentrations are maintained longer.
The 40 mg daily subcutaneous dose was selected based on Phase 2 dose-ranging data showing it was well tolerated and produced consistent pharmacodynamic effects. While higher doses have not been extensively studied in long-term trials, the dose-response data suggest that the 40 mg dose approaches the plateau of the efficacy curve, meaning that increasing the dose further would provide diminishing incremental benefit while potentially increasing injection site reactions.
Comparison to Other Mitochondrial Targeting Strategies
SS-31 is not the only molecule designed to target mitochondria, but it occupies a unique position in the field. Triphenylphosphonium (TPP+)-conjugated compounds, such as MitoQ (a coenzyme Q10 analog attached to a TPP+ moiety), rely on membrane potential for accumulation. While effective in cells with intact membrane potential, they lose their targeting advantage in damaged mitochondria. They also accumulate to potentially toxic levels in healthy, fully polarized mitochondria, creating a narrow therapeutic window.
Mitochondria-penetrating peptides (MPPs) represent another class of targeting molecules, but most are designed primarily for cargo delivery rather than direct therapeutic activity. SS-31 is unique in that the targeting moiety and the therapeutic moiety are one and the same: the peptide that reaches the inner membrane is also the molecule that stabilizes cardiolipin.
SkQ1 (plastoquinonyl-decyl-triphenylphosphonium), developed by Vladimir Skulachev's group in Russia, is another mitochondria-targeted antioxidant that has reached clinical development for ophthalmic applications. Like MitoQ, it relies on TPP+ for targeting and membrane potential for accumulation, sharing the same potential-dependence limitation.
The unique characteristics of SS-31's targeting mechanism, including its potential-independence, its direct interaction with cardiolipin, and its complex effects on ETC function, distinguish it from all other mitochondrial-targeting approaches currently in development. These properties explain why SS-31 has advanced further in clinical development than any competing mitochondrial-targeted therapeutic.
Tissue-Specific Uptake and Distribution
While SS-31 concentrates in mitochondria across all cell types tested, the absolute uptake varies by tissue due to differences in mitochondrial density, cardiolipin content, and blood flow. Tissues with the highest mitochondrial density, including cardiac muscle, skeletal muscle, renal proximal tubules, retinal pigment epithelium, and neurons, show the greatest absolute uptake of SS-31. This natural distribution pattern is therapeutically advantageous, as these are precisely the tissues most vulnerable to mitochondrial dysfunction and most relevant to the clinical conditions being studied.
Studies using radiolabeled SS-31 in rodents have mapped the tissue distribution following both intravenous and subcutaneous administration. After subcutaneous injection, the peptide is rapidly absorbed into the systemic circulation and distributed to all major organs. The heart, kidneys, and skeletal muscle show particularly high uptake relative to plasma concentrations. Brain uptake is lower in absolute terms, reflecting the blood-brain barrier, but still reaches pharmacologically relevant concentrations, supporting the neuroprotective effects observed in preclinical studies.
The subcellular distribution within each tissue confirms the mitochondrial specificity. Using immunogold electron microscopy and fluorescent analogs, researchers have shown that SS-31 localizes primarily to the inner mitochondrial membrane, with minimal presence in the outer membrane, intermembrane space, or matrix. This precise localization positions the peptide at the exact site where cardiolipin resides and where the electron transport chain operates.
Time Course of Mitochondrial Effects
One of the most striking features of SS-31 is the rapid onset of its mitochondrial effects. In isolated mitochondria, improvements in respiratory function can be measured within minutes of SS-31 exposure. In intact cells, increases in ATP production are detectable within 15-30 minutes. In whole animals, improvements in cardiac or muscle function are measurable within 1-4 hours of systemic administration.
This rapid onset contrasts with most other interventions that target mitochondrial function. Strategies based on mitochondrial biogenesis (such as exercise training or PGC-1alpha activation) require days to weeks to increase mitochondrial content. Coenzyme Q10 supplementation shows very gradual effects due to slow mitochondrial incorporation. And gene therapy approaches for mitochondrial diseases are still in early development. SS-31's ability to improve existing mitochondrial function within minutes of reaching its target provides a unique therapeutic profile well suited to both acute conditions (like ischemia-reperfusion) and chronic conditions (where daily dosing maintains continuous mitochondrial support).
The duration of effect after a single dose depends on the tissue context. In plasma, SS-31 has a half-life of approximately 2-3 hours. But within mitochondrial membranes, the peptide's residence time is considerably longer, likely because its binding to cardiolipin creates a reservoir that is not rapidly depleted by systemic clearance. This pharmacokinetic-pharmacodynamic dissociation, where the biological effect outlasts the plasma concentration, is characteristic of drugs with high tissue affinity and supports the once-daily dosing regimen used in chronic clinical trials.
Cardiolipin Stabilization
Cardiolipin is the defining lipid of mitochondria, found almost exclusively in the inner mitochondrial membrane where it constitutes roughly 20% of total lipid content. SS-31's therapeutic mechanism centers on stabilizing this lipid, making cardiolipin biology essential to understanding how the peptide works and why its effects span multiple organ systems and diseases.
What is Cardiolipin and Why Does It Matter?
Cardiolipin (1,3-bis(sn-3'-phosphatidyl)-sn-glycerol) is a phospholipid unlike any other in the cell. Its dimeric structure, consisting of two phosphatidic acid molecules joined through a central glycerol, gives it four fatty acyl chains and two phosphate headgroups. This unique architecture allows cardiolipin to assume cone-shaped conformations that promote the curvature necessary for cristae formation, the deep infoldings of the inner mitochondrial membrane that dramatically increase the surface area available for oxidative phosphorylation.
Beyond its structural role, cardiolipin functions as an essential co-factor for virtually every major protein complex in the inner mitochondrial membrane. Crystal structures of Complexes III and IV show cardiolipin molecules tightly bound within the protein structures, where they participate directly in proton translocation and electron transfer. ATP synthase (Complex V) similarly requires cardiolipin for optimal activity. The adenine nucleotide translocator (ANT), the most abundant protein in the inner mitochondrial membrane, has multiple cardiolipin binding sites that regulate its transport activity.
The functional consequences of cardiolipin loss or modification are severe. When cardiolipin content decreases, or when its acyl chains become oxidized, the efficiency of oxidative phosphorylation declines precipitously. Cristae structure deteriorates. Supercomplexes disassemble. ROS production increases. And ultimately, the release of cytochrome c from the outer surface of the inner membrane triggers the mitochondrial apoptosis pathway, leading to programmed cell death.
Cardiolipin Remodeling: The Tafazzin Connection
In healthy cells, cardiolipin undergoes constant remodeling to maintain the specific acyl chain composition required for optimal function. In most tissues, mature cardiolipin is enriched in linoleoyl (18:2) acyl chains, creating a highly symmetric tetralinoleoyl cardiolipin (L4CL) that is thought to provide the optimal lipid environment for respiratory chain activity.
The enzyme tafazzin (encoded by the TAFAZZIN gene, also known as TAZ) is the primary transacylase responsible for cardiolipin remodeling. It catalyzes the exchange of acyl chains between cardiolipin and other phospholipids, progressively enriching cardiolipin in the preferred linoleoyl species. Mutations in the TAFAZZIN gene cause Barth syndrome, a rare X-linked genetic disorder characterized by cardiomyopathy, skeletal myopathy, neutropenia, and growth retardation.
In Barth syndrome, the absence of functional tafazzin leads to dramatic alterations in cardiolipin profile: total cardiolipin content decreases, the remaining cardiolipin has aberrant acyl chain composition, and monolysocardiolipin (MLCL), a degradation product normally present in trace amounts, accumulates. The MLCL-to-CL ratio serves as a diagnostic biomarker for Barth syndrome and as a pharmacodynamic marker in clinical trials of elamipretide.
The connection between Barth syndrome and SS-31 is direct. By binding to whatever cardiolipin remains in tafazzin-deficient mitochondria, SS-31 can partially compensate for the structural and functional deficits caused by abnormal cardiolipin composition. This doesn't correct the underlying genetic defect, but it can stabilize mitochondrial function enough to produce clinically meaningful improvements in muscle strength and exercise capacity.
Cardiolipin Oxidation in Disease and Aging
While Barth syndrome represents the most dramatic example of cardiolipin pathology, cardiolipin oxidation occurs in a wide range of common diseases. The inner mitochondrial membrane, where cardiolipin resides, is the primary site of cellular ROS production. The proximity of cardiolipin's polyunsaturated acyl chains (particularly the bis-allylic hydrogens of linoleic acid) to the electron transport chain makes them highly vulnerable to oxidative attack.
In cardiac ischemia-reperfusion injury, the burst of ROS production that accompanies reperfusion triggers massive cardiolipin peroxidation. This disrupts respiratory chain function, promotes cytochrome c release, and drives cardiomyocyte death. The extent of cardiolipin oxidation correlates directly with infarct size and functional impairment. SS-31 administered before or during reperfusion can prevent this cardiolipin oxidation cascade, reducing infarct size in preclinical models by 40-60%.
In heart failure, chronic neurohormonal activation and oxidative stress lead to progressive cardiolipin depletion and remodeling. Studies of explanted human hearts from patients with end-stage heart failure have shown significant reductions in total cardiolipin content and alterations in acyl chain composition compared to non-failing hearts. Critically, Sabbah and colleagues (2016) demonstrated that elamipretide normalized cardiolipin content and restored mitochondrial function in dogs with experimentally induced heart failure, supporting the concept that cardiolipin stabilization can address not just the symptoms but a root cause of cardiac dysfunction.
In the aging process itself, cardiolipin content decreases progressively across multiple tissues. Studies in aged rodents have documented reductions of 30-50% in cardiac, skeletal muscle, hepatic, and cerebral cardiolipin levels compared to young animals. This age-related cardiolipin loss tracks closely with declines in mitochondrial function, increases in ROS production, and deterioration of tissue function. The ability of SS-31 to reverse age-related mitochondrial dysfunction in animal models correlates with its effects on cardiolipin-dependent processes, suggesting that age-related cardiolipin loss may be a treatable condition rather than an inevitable consequence of biological aging.
SS-31's Cardiolipin Stabilization: Molecular Details
The precise molecular mechanism by which SS-31 stabilizes cardiolipin involves several complementary actions. First, by binding at the phospholipid headgroup-acyl chain interface, SS-31 physically shields the vulnerable bis-allylic hydrogens from attack by ROS. This is not a stoichiometric antioxidant reaction (where one molecule of antioxidant neutralizes one ROS); instead, it's a structural protection that reduces the accessibility of the oxidation-sensitive sites.
Second, SS-31 modulates the conformation of cardiolipin in ways that favor its functional interactions with respiratory chain proteins. Molecular dynamics simulations have shown that SS-31 promotes the cone-shaped conformation of cardiolipin that supports cristae curvature and supercomplex assembly, while discouraging the lamellar conformations associated with membrane flattening and functional impairment.
Third, SS-31 prevents the cardiolipin-cytochrome c interaction that catalyzes cardiolipin peroxidation. When cytochrome c binds to cardiolipin in the absence of SS-31, the protein's heme group can catalyze the oxidation of cardiolipin's acyl chains, creating a self-amplifying damage cycle. SS-31 competes with this deleterious binding mode, keeping cytochrome c in its electron-carrier conformation and preventing the initiation of the peroxidation cascade.
Fourth, by maintaining the structural integrity of the inner mitochondrial membrane, SS-31 preserves the proton gradient (delta psi and delta pH) that drives ATP synthesis. Cardiolipin oxidation leads to increased proton leak across the inner membrane, dissipating the proton motive force and reducing the efficiency of ATP production. SS-31 reduces this leak, ensuring that a higher proportion of the energy from electron transport is captured as ATP rather than lost as heat.
Implications for Therapeutic Applications
The centrality of cardiolipin to mitochondrial function explains why SS-31's therapeutic effects extend across so many different conditions. Every tissue in the body depends on mitochondrial ATP production, and every mitochondrion depends on cardiolipin for optimal function. When cardiolipin is damaged, whether by genetic mutations (Barth syndrome), acute oxidative stress (ischemia-reperfusion), chronic disease (heart failure), or biological aging, the downstream consequences follow the same pattern: impaired electron transport, increased ROS, decreased ATP, and eventual cell death.
SS-31 intervenes at this convergence point. By stabilizing cardiolipin regardless of why it became destabilized, the peptide addresses a common upstream cause rather than a disease-specific downstream symptom. This mechanism explains both the breadth of SS-31's preclinical efficacy and the rationale for testing it across multiple clinical indications. Other compounds that support mitochondrial health through different mechanisms, such as NAD+ (which supports mitochondrial enzyme function) and MOTS-c (a mitochondria-derived peptide that regulates metabolic homeostasis), may complement SS-31's membrane-level effects, potentially offering multi-pathway approaches to mitochondrial restoration.
Cardiolipin in Different Organ Systems
The importance of cardiolipin varies somewhat by organ system, reflecting differences in mitochondrial density and metabolic demands. Understanding these tissue-specific roles helps explain why SS-31 produces different clinical effects in different conditions.
In the heart, cardiolipin is particularly critical because of the organ's extraordinarily high ATP demand. Cardiac mitochondria contain the highest cardiolipin concentrations of any tissue, and even modest reductions in cardiolipin content produce disproportionate declines in cardiac function. The heart's continuous contractile activity means it cannot tolerate even brief periods of bioenergetic failure, making it acutely sensitive to cardiolipin perturbation. Heart failure, cardiac aging, and ischemia-reperfusion injury all involve cardiolipin pathology.
In skeletal muscle, cardiolipin supports the metabolic flexibility required for transitions between rest and exercise. During exercise, skeletal muscle ATP demand can increase 100-fold above resting levels, requiring maximum efficiency from the oxidative phosphorylation machinery. Cardiolipin depletion in aged or diseased muscle impairs this capacity, contributing to exercise intolerance, fatigue, and sarcopenia. The rapid improvements in muscle function observed with SS-31 in aged animals reflect the restoration of cardiolipin-dependent respiratory capacity.
In the kidney, the proximal tubular cells are among the most metabolically active in the body, reabsorbing over 99% of the filtered sodium through ATP-dependent pumps. These cells rely almost entirely on oxidative phosphorylation (rather than glycolysis) for their energy needs, making them extremely sensitive to cardiolipin loss. Acute kidney injury, diabetic nephropathy, and age-related kidney decline all involve mitochondrial dysfunction in the proximal tubule.
In the retina, the retinal pigment epithelium (RPE) has one of the highest oxidative metabolic rates in the body, supporting the continuous regeneration of photoreceptor outer segments. RPE mitochondria are subjected to intense oxidative stress from light exposure and high oxygen tension, making their cardiolipin particularly vulnerable to peroxidation. The accumulation of lipofuscin and drusen in age-related macular degeneration correlates with declining RPE mitochondrial function.
In the brain, neurons have high energy demands for maintaining membrane potentials, synthesizing neurotransmitters, and supporting synaptic transmission. While the blood-brain barrier limits drug delivery, SS-31 reaches brain mitochondria at pharmacologically active concentrations and has shown neuroprotective effects in multiple preclinical models, from stroke to Parkinson's disease to age-related cognitive decline.
Cardiolipin as a Therapeutic Target: Current and Future Approaches
SS-31 is not the only approach to targeting cardiolipin, though it is the most clinically advanced. Other strategies under investigation include genetic approaches to augment tafazzin function (particularly relevant for Barth syndrome), enzyme replacement strategies, small molecules that promote cardiolipin synthesis, and dietary interventions that influence cardiolipin acyl chain composition (such as linoleic acid supplementation).
The success of SS-31 in reaching FDA approval validates cardiolipin as a druggable target and may catalyze the development of next-generation cardiolipin-targeting compounds. Potential improvements over SS-31 could include oral bioavailability (eliminating the need for daily injections), longer duration of action, tissue selectivity (concentrating effects in specific organs), and reduced injection site reactions.
Meanwhile, compounds that support overall mitochondrial health through complementary mechanisms continue to generate interest. NAD+ precursors restore the coenzyme pool needed for mitochondrial enzyme function. 5-Amino-1MQ targets NNMT to boost cellular NAD+ levels. MOTS-c activates AMPK signaling to improve metabolic homeostasis. And Humanin, a mitochondria-derived peptide, protects against stress-induced cell death. These compounds address different aspects of the mitochondrial dysfunction equation, and their potential for combined use with SS-31 represents an active area of investigation in the biohacking and longevity communities.
Heart Failure Clinical Trials
Heart failure affects over 64 million people worldwide and remains a leading cause of morbidity and mortality despite decades of pharmacological advances. Mitochondrial dysfunction has been identified as both a cause and consequence of cardiac failure, making SS-31's cardiolipin-stabilizing mechanism particularly relevant to this condition. Multiple clinical trials have evaluated elamipretide in cardiac settings with mixed but informative results.
Rationale: Why Target Mitochondria in Heart Failure?
The heart is the most metabolically active organ in the body, consuming approximately 6 kilograms of ATP per day to sustain its continuous contractile function. Cardiomyocytes are packed with mitochondria, which occupy roughly 30% of cell volume, the highest mitochondrial density of any cell type. This extreme dependence on oxidative phosphorylation makes the heart uniquely vulnerable to mitochondrial dysfunction.
In heart failure, mitochondrial impairment is well documented. Studies of both animal models and explanted human hearts have consistently shown reduced mitochondrial respiratory capacity, decreased ATP production, increased ROS generation, cardiolipin depletion and remodeling, and disrupted cristae architecture. These changes are not merely correlative; interventions that restore mitochondrial function can improve cardiac contractility and reverse ventricular remodeling in preclinical models.
Conventional heart failure therapies (ACE inhibitors, beta-blockers, angiotensin receptor-neprilysin inhibitors, SGLT2 inhibitors, and mineralocorticoid receptor antagonists) target neurohormonal pathways that drive disease progression. While effective, they don't directly address the underlying bioenergetic deficit. This gap in the therapeutic arsenal provided the rationale for testing elamipretide, which could potentially restore cardiac energetics at the mitochondrial level.
Early Phase Trial: Proof of Concept
The initial human cardiac trial was a double-blind, placebo-controlled, ascending-dose study in patients with stable heart failure with reduced ejection fraction (HFrEF, defined as LVEF of 35% or below). Patients received escalating 4-hour intravenous infusions of elamipretide at doses of 0.005, 0.05, and 0.25 mg/kg/hr, with echocardiographic assessment before, during, and after infusion.
The results provided the first evidence of cardiac effects in humans. In the highest dose cohort (0.25 mg/kg/hr), compared with placebo, there was a significant decrease in left ventricular end-diastolic volume (-18 mL, P = 0.009) and left ventricular end-systolic volume (-14 mL, P = 0.005) at the end of infusion. These volume reductions, which indicate improved cardiac contractility and reduced ventricular dilation, correlated with peak plasma concentrations of elamipretide. The treatment was well tolerated, with no significant adverse events reported.
While small in scale (36 patients total), this trial established that elamipretide could produce measurable improvements in cardiac function within hours of administration, consistent with its rapid mitochondrial uptake and mechanism of action. The magnitude of volume reduction, while modest in absolute terms, was clinically relevant and supported advancement to larger trials.
EMBRACE-STEMI: Acute Myocardial Infarction
The EMBRACE-STEMI trial (Evaluation of Myocardial Effects of Bendavia for Reducing Reperfusion Injury in Patients with Acute Coronary Events - ST-Segment Elevation Myocardial Infarction) addressed a different cardiac scenario: could SS-31 reduce damage from reperfusion injury when administered during primary percutaneous coronary intervention (PCI) for acute heart attack?
The trial enrolled 118 patients with anterior ST-elevation myocardial infarction undergoing primary PCI. Patients were randomized to receive a 1-hour intravenous infusion of elamipretide (0.05 mg/kg/hr) or placebo, initiated prior to PCI and continued during the procedure. The primary endpoint was myocardial infarct size assessed by cardiac magnetic resonance imaging (CMR) at 72 hours post-PCI.
The primary endpoint was not met: elamipretide did not significantly reduce infarct size measured by CMR at 72 hours compared to placebo. However, a secondary analysis revealed that elamipretide treatment was associated with a reduced incidence of new-onset heart failure within 24 hours of PCI (2.5% vs. 14.3% in the placebo group, P = 0.03). This finding, while exploratory, suggested that elamipretide's protective effects might manifest in functional outcomes rather than in the anatomical endpoint of infarct size.
Several factors may have contributed to the neutral primary endpoint. The relatively low dose used (based on early pharmacokinetic data) may have been insufficient for maximal cardioprotection. The timing of administration, which began shortly before PCI, left limited time for mitochondrial loading before the reperfusion event. And the 72-hour CMR endpoint, while well-validated, may not have captured the full spectrum of elamipretide's protective effects, some of which might manifest over longer timeframes through preserved mitochondrial function and reduced ongoing cell death.
PROGRESS-HF: Chronic Heart Failure
The PROGRESS-HF trial was a multicenter, randomized, double-blind, placebo-controlled Phase 2 study designed to evaluate the efficacy of subcutaneous elamipretide in patients with chronic HFrEF. Seventy-one patients with stable heart failure and LVEF of 40% or below were randomized to receive placebo, 4 mg, or 40 mg of elamipretide subcutaneously once daily for 28 consecutive days.
The primary endpoint was change in left ventricular end-systolic volume (LVESV) from baseline to Week 4, assessed by CMR. The trial did not meet its primary endpoint: the change in LVESV was not significantly different between the elamipretide groups and placebo at 4 weeks.
However, the trial provided several valuable insights. First, subcutaneous elamipretide was well tolerated at both dose levels, with injection site reactions being the most common treatment-emergent adverse event. There were no significant safety signals, supporting the feasibility of chronic daily subcutaneous administration. Second, exploratory analyses suggested trends toward improvement in some secondary endpoints, particularly in the 40 mg group, though none reached statistical significance in this small trial.
The negative primary result in PROGRESS-HF raises important questions about the clinical development of elamipretide for heart failure. A 4-week treatment duration may have been too short to observe meaningful reverse remodeling in a chronic condition that develops over months to years. The sample size of 71 patients, while appropriate for a Phase 2 study, limited statistical power to detect modest but clinically relevant treatment effects. And LVESV, while an important surrogate marker, may not have been the most sensitive endpoint for detecting improvements in mitochondrial function.
Mechanistic Studies in Human Heart Tissue
Complementing the clinical trials, important mechanistic data has come from studies of elamipretide in human cardiac tissue. Sabbah and colleagues (2016) published a key study demonstrating that elamipretide improved mitochondrial function in failing human hearts. Using permeabilized muscle fibers from explanted hearts of patients with end-stage heart failure, they showed that acute exposure to elamipretide significantly increased maximal mitochondrial respiration and ATP production.
The same group later showed that elamipretide restored mitochondrial function, normalized cardiolipin content, and improved left ventricular function in dogs with experimentally induced heart failure over a 3-month treatment period. These results supported the concept that longer treatment durations might be necessary to observe clinical benefits and that the cardiolipin-stabilizing mechanism translated from preclinical models to human cardiac tissue.
Future Directions in Cardiac Applications
Despite the mixed results from completed trials, the rationale for targeting mitochondria in heart failure remains strong. The preclinical evidence is consistent and compelling: mitochondrial dysfunction drives cardiac failure, and cardiolipin stabilization can restore cardiac bioenergetics. The disconnect between preclinical promise and clinical results likely reflects the challenges of translational medicine, including dose optimization, endpoint selection, treatment duration, and patient selection.
Future clinical programs in heart failure may benefit from several refinements. Longer treatment durations (3-6 months or more) would allow time for mitochondrial restoration to translate into structural reverse remodeling. Biomarker-guided patient selection could identify patients with the greatest degree of mitochondrial dysfunction and therefore the highest likelihood of benefit. And functional endpoints, such as exercise capacity, quality of life, and event-free survival, may capture the full clinical impact of improved mitochondrial function more effectively than imaging-based volume endpoints.
For individuals interested in the broader field of cardiac therapeutics, semaglutide has also shown cardiovascular benefits in large clinical trials, acting through entirely different mechanisms (GLP-1 receptor agonism, weight reduction, anti-inflammatory effects). The convergence of multiple therapeutic approaches targeting different aspects of cardiac disease represents a promising direction for patients who haven't responded adequately to conventional therapy. The GLP-1 research hub covers the cardiovascular data for incretin-based therapies.
Mitochondrial ATP Production After SS-31 Treatment
Data from preclinical studies showing the restoration of mitochondrial ATP production in aged animals treated with SS-31, expressed as percentage of young healthy baseline.
Lessons Learned from Cardiac Trials
The cardiac trial program for elamipretide provides instructive lessons for the broader field of mitochondrial therapeutics. Several themes emerge from analyzing the pattern of results across the different cardiac trials.
First, treatment duration matters enormously. The early-phase IV infusion trial showed acute effects within hours, but these didn't translate into sustained improvement in the 4-week PROGRESS-HF study. In contrast, the TAZPOWER extension in Barth syndrome showed progressive improvement over 168 weeks. Cardiac remodeling is a slow process, and reversing years of mitochondrial damage and structural deterioration likely requires months to years of sustained treatment rather than weeks.
Second, endpoint selection critically influences trial outcomes. LVESV, the primary endpoint in PROGRESS-HF, is a well-validated surrogate marker for cardiac outcomes, but it may not be the most sensitive measure of mitochondrial-driven improvement. Cardiac energetics (measured by 31P-MRS), exercise capacity, quality of life, or composite clinical endpoints might better capture the full spectrum of benefit from improved mitochondrial function.
Third, patient selection should consider the degree of underlying mitochondrial dysfunction. Patients with the most severe mitochondrial impairment may have the most to gain from cardiolipin stabilization, while those with relatively preserved mitochondrial function may show smaller incremental benefits. Biomarker-guided enrollment using measures of mitochondrial function (such as 31P-MRS or circulating biomarkers of mitochondrial stress) could enrich trial populations for likely responders.
Fourth, the dose-response relationship needs further exploration. The doses used in cardiac trials were based on early pharmacokinetic data and may not have been optimized for cardiac mitochondrial loading. Higher doses, longer loading periods, or alternative dosing regimens might produce more consistent cardiac benefits.
These lessons apply not only to elamipretide but to the entire class of mitochondrial-targeted therapeutics. As the field matures, trial designs will likely evolve to incorporate longer treatment durations, functional endpoints, biomarker-guided enrollment, and optimized dosing strategies. The cardiac indication remains compelling based on preclinical data, and future trials incorporating these refinements may yield more definitive results.
For those interested in cardiac health more broadly, the cardiovascular benefits of semaglutide and tirzepatide have been established through large-scale outcome trials with tens of thousands of patients, including the SELECT trial for semaglutide which showed a 20% reduction in major adverse cardiovascular events. The GLP-1 research hub provides detailed coverage of these cardiovascular findings.
Barth Syndrome Research

Barth syndrome is a rare, X-linked genetic disorder caused by mutations in the TAFAZZIN gene, which encodes the enzyme responsible for cardiolipin remodeling. It provided the proving ground for elamipretide's clinical development and the indication for which the peptide achieved its first FDA approval in September 2025.
Understanding Barth Syndrome
Barth syndrome (BTHS) affects approximately 1 in 300,000 to 1 in 400,000 live births, overwhelmingly in males due to its X-linked recessive inheritance pattern. The disease is caused by loss-of-function mutations in the TAFAZZIN gene located on the X chromosome. The tafazzin enzyme is a phospholipid transacylase that remodels cardiolipin by exchanging its acyl chains, maintaining the linoleoyl-rich composition required for optimal mitochondrial function.
Without functional tafazzin, cardiolipin composition becomes aberrant. Total cardiolipin levels decrease, the normally predominant tetralinoleoyl species is depleted, and monolysocardiolipin (MLCL) accumulates. The MLCL-to-CL ratio, which is normally very low (less than 0.1), increases dramatically in Barth syndrome patients, often exceeding 1.0. This ratio serves as both a diagnostic marker and a pharmacodynamic endpoint in clinical trials.
The clinical manifestations of Barth syndrome reflect the widespread consequences of cardiolipin deficiency. Cardiomyopathy, typically dilated or left ventricular noncompaction, appears in infancy or early childhood and is the leading cause of mortality. Skeletal myopathy causes weakness, exercise intolerance, and delayed motor development. Cyclical neutropenia increases susceptibility to bacterial infections. And growth retardation affects height and lean body mass throughout development.
Before elamipretide, treatment for Barth syndrome was entirely supportive: heart failure medications for cardiomyopathy, granulocyte-colony stimulating factor (G-CSF) for neutropenia, nutritional support for growth, and physical therapy for myopathy. No approved therapy addressed the underlying mitochondrial dysfunction. This unmet medical need made Barth syndrome a natural target for elamipretide development.
The TAZPOWER Trial: Design and Initial Results
TAZPOWER was a Phase 2/3 clinical trial designed to evaluate the safety and efficacy of elamipretide in patients with Barth syndrome. The trial consisted of two phases: a 12-week randomized, double-blind, placebo-controlled crossover period, followed by a 168-week open-label extension (OLE) in which all patients received elamipretide 40 mg subcutaneously once daily.
Twelve patients (ages 12 and older) with genetically confirmed Barth syndrome were enrolled in the double-blind phase. The primary endpoint was the change in 6-minute walk test (6MWT) distance from baseline to Week 12. Secondary endpoints included the BTHS Symptom Assessment (BTHS-SA) Total Fatigue score, shuttle walk and run tests, and muscle strength measures.
The initial 12-week double-blind phase did not meet its primary or secondary endpoints. The 6MWT distance improvement with elamipretide was not statistically different from placebo at the 12-week timepoint. This result was disappointing but not entirely unexpected, given the small sample size (inherent to studying a rare disease affecting approximately 150 individuals in the United States), the crossover design (which can complicate interpretation when treatment effects persist beyond the washout period), and the relatively short treatment duration.
TAZPOWER Open-Label Extension: Long-Term Results
The open-label extension phase of TAZPOWER provided substantially more encouraging data. Of the 12 patients who completed the double-blind phase, 10 entered the open-label extension and continued daily subcutaneous elamipretide. Eight patients reached the Week 168 (3.2-year) assessment.
The 168-week open-label extension results, published in 2024, demonstrated:
- 6-Minute Walk Test: Significant improvements from OLE baseline occurred at all assessment timepoints, with a cumulative improvement of 96.1 meters at Week 168 (P = 0.003). This magnitude of improvement is clinically meaningful, as a 30-50 meter improvement in 6MWT distance is generally considered the minimum clinically important difference in cardiac and respiratory diseases.
- Fatigue Scores: Mean BTHS-SA Total Fatigue scores improved (decreased) from OLE baseline at all timepoints throughout the extension period, indicating sustained reduction in the subjective experience of fatigue.
- Cardiac Function: Three-dimensional echocardiographic assessment showed improvements in left ventricular stroke volume, end-diastolic volume, and end-systolic volume, with significant trends for improvement from baseline to Week 168.
- Biomarker Response: MLCL/CL ratio values showed improvement over the extension period, supporting the mechanistic hypothesis that elamipretide improves cardiolipin metabolism.
- Muscle Strength: Knee extensor muscle strength improved from baseline, providing the efficacy endpoint that ultimately supported FDA approval.
Natural History Comparison Study
A separate natural history comparison study provided additional context for the TAZPOWER results. Researchers compared the functional trajectories of TAZPOWER participants to those of untreated Barth syndrome patients followed in natural history registries. The comparison showed that elamipretide-treated patients maintained or improved their functional capacity over 3+ years, while untreated patients showed progressive decline in walking ability and muscle function over comparable periods.
This comparison was particularly informative because it addressed a key limitation of the open-label extension design: without a concurrent control group, improvements could theoretically be attributed to placebo effects, practice effects, or natural disease fluctuation. The natural history data showed that untreated Barth syndrome is a progressive condition, making sustained improvements in the treated group all the more meaningful.
FDA Approval: FORZINITY
On September 19, 2025, the FDA granted accelerated approval to FORZINITY (elamipretide) injection for the improvement of muscle strength in adult and pediatric patients with Barth syndrome weighing at least 30 kilograms. The approval was based on the knee extensor muscle strength data from the TAZPOWER open-label extension.
Key aspects of the approval include:
- Accelerated approval pathway: The approval was based on a surrogate endpoint (muscle strength) reasonably likely to predict clinical benefit. Continued approval is contingent upon verification of clinical benefit in confirmatory trials.
- Dosing: The approved dose is 40 mg administered subcutaneously once daily.
- Weight restriction: The label specifies patients weighing at least 30 kg. Stealth BioTherapeutics is working with the FDA to generate data for expanding the indication to smaller children.
- First treatment for Barth syndrome: FORZINITY is the first FDA-approved medication specifically indicated for Barth syndrome.
- First mitochondria-targeted therapeutic: The approval represents a broader milestone for mitochondrial medicine, validating the concept that targeting mitochondrial membrane biology can produce clinically meaningful benefits.
Preclinical Barth Syndrome Research
Complementing the clinical data, preclinical studies in Barth syndrome models have provided mechanistic insights. A 2024 study published in Scientific Reports demonstrated that SS-31 treatment restored mitochondrial morphology and corrected defective mitophagy in a murine model of Barth syndrome. The tafazzin-knockdown mice showed fragmented mitochondria with disorganized cristae, decreased expression of mitophagy-related proteins, and accumulation of damaged mitochondria. SS-31 treatment normalized mitochondrial morphology, restored expression of proteins involved in mitochondrial dynamics (fusion and fission), and reactivated mitophagy pathways, allowing for the selective removal of irreparably damaged mitochondria.
These findings expanded the understanding of how SS-31 works in Barth syndrome beyond simple cardiolipin stabilization. By restoring the quality control mechanisms that maintain a healthy mitochondrial population, elamipretide may prevent the accumulation of dysfunctional mitochondria that drives progressive organ damage in Barth syndrome.
Barth Syndrome: Detailed Clinical Presentation and Diagnosis
Understanding the clinical presentation of Barth syndrome is essential for appreciating why elamipretide's approval was so significant. The condition typically presents in infancy or early childhood, though the diagnosis is often delayed because the symptoms overlap with many other conditions.
The cardiac manifestations are usually the first to attract clinical attention. Dilated cardiomyopathy, characterized by enlarged and weakened heart chambers, develops in the majority of patients by age 2. Left ventricular noncompaction cardiomyopathy, a distinctive pattern of excessive trabeculation in the ventricular myocardium, is also common and may coexist with dilated cardiomyopathy. Some patients present with neonatal cardiomyopathy severe enough to require cardiac transplantation in the first year of life. Others maintain relatively stable cardiac function through childhood but develop progressive heart failure in adolescence or adulthood. The variability in cardiac phenotype, even among patients with the same TAFAZZIN mutation, suggests that modifier genes and environmental factors influence disease severity.
Skeletal myopathy in Barth syndrome is characterized by proximal muscle weakness, delayed motor milestones, exercise intolerance, and generalized fatigue. Boys with Barth syndrome typically learn to walk later than their peers and have difficulty with activities requiring sustained physical effort. The myopathy is not progressive in the same way as muscular dystrophies; rather, it represents a chronic limitation in the maximum force and endurance that muscles can achieve, directly traceable to mitochondrial bioenergetic failure. The 6-minute walk test and measures of knee extensor strength used in the TAZPOWER trial were specifically chosen to capture these functional limitations.
Cyclical neutropenia affects approximately 90% of Barth syndrome patients and contributes to recurrent and sometimes serious bacterial infections. The neutropenia is often severe (absolute neutrophil count below 500 cells/microL) and follows an unpredictable cycling pattern, making prophylactic management challenging. Treatment with granulocyte colony-stimulating factor (G-CSF) can boost neutrophil counts during nadir periods, but the response is variable. The mechanism linking tafazzin deficiency to neutropenia involves impaired neutrophil differentiation and survival, likely related to mitochondrial dysfunction in myeloid precursor cells.
Growth retardation affects both height and lean body mass. Boys with Barth syndrome tend to be shorter than their age-matched peers and have reduced muscle mass relative to body size. The growth delay is not due to growth hormone deficiency but rather to the metabolic consequences of chronic mitochondrial dysfunction: reduced ability to synthesize new tissue (an energy-intensive process) combined with increased resting metabolic demands from inefficient mitochondria.
Diagnosis of Barth syndrome involves a combination of clinical suspicion, biochemical testing, and genetic confirmation. The MLCL-to-CL ratio, measured in blood spots or cultured fibroblasts, is elevated in virtually all cases and serves as a reliable screening test. Definitive diagnosis requires identification of a pathogenic variant in the TAFAZZIN gene by DNA sequencing. Newborn screening for Barth syndrome is not currently available in most jurisdictions, contributing to diagnostic delays that average 3-5 years from symptom onset.
Interpreting the TAZPOWER Results: Statistical and Clinical Considerations
The mixed results from the TAZPOWER trial, with a negative blinded phase followed by a positive open-label extension, deserve careful interpretation. Several factors explain this apparent discrepancy.
The 12-week blinded phase enrolled only 12 patients in a crossover design, giving it very limited statistical power. With such a small sample, even a real treatment effect of moderate size has a low probability of reaching statistical significance. The crossover design, while efficient for some pharmacological questions, introduces additional complexity: if elamipretide's effects persist beyond the washout period (which is plausible for a drug that stabilizes mitochondrial membranes), residual benefit from the first treatment period could attenuate the apparent effect in the second period.
The 12-week treatment duration may also have been insufficient for maximal benefit. The TAZPOWER OLE data showed that improvements continued to accumulate throughout the 168-week extension, with the 6MWT distance improving progressively at each assessment timepoint. At 12 weeks, patients may have been on the ascending portion of a dose-response curve that required months or years to reach plateau.
The open-label extension, while unblinded, provides complementary evidence. The lack of a concurrent control group is a limitation, but the natural history comparison data mitigates this concern. Untreated Barth syndrome patients show progressive functional decline, not stability or improvement. The fact that treated patients showed significant improvement against this expected trajectory of decline provides meaningful evidence of efficacy, even in the absence of formal blinding.
The FDA's decision to grant accelerated approval based on the muscle strength data from the OLE reflects a regulatory framework designed to address the needs of patients with serious conditions lacking adequate treatment options. The accelerated pathway allows approval based on surrogate endpoints that are reasonably likely to predict clinical benefit, with post-marketing confirmatory trials required to verify that the surrogate translates into real clinical improvement.
Patient Perspective and Impact
For the Barth syndrome community, the approval of FORZINITY represented the culmination of decades of advocacy, research collaboration, and patient participation in clinical trials. The Barth Syndrome Foundation played a key role in patient identification, trial recruitment, and regulatory advocacy throughout elamipretide's development.
Living with Barth syndrome means navigating daily challenges: fatigue that limits activity, cardiac complications that require ongoing monitoring, immune system vulnerabilities, and growth delays. The availability of a treatment that addresses the root mitochondrial cause, rather than just managing symptoms, represents a fundamental shift in the treatment paradigm. While elamipretide doesn't cure Barth syndrome, the improvements in muscle strength, walking distance, and cardiac function documented in TAZPOWER translate into tangible improvements in daily functioning and quality of life.
For clinicians managing patients with mitochondrial disorders, the Barth syndrome approval also establishes a precedent. It demonstrates that mitochondrial dysfunction can be pharmacologically targeted and that regulatory agencies will consider approval of therapies based on functional outcomes in rare mitochondrial diseases. This precedent may facilitate the development of treatments for other mitochondrial disorders, a group of conditions that collectively affect an estimated 1 in 5,000 individuals.
Age-Related Mitochondrial Dysfunction
Mitochondrial dysfunction is one of the nine recognized hallmarks of aging, contributing to the progressive decline in tissue function that characterizes biological aging across virtually every organ system. SS-31's ability to restore mitochondrial function in aged tissues has generated substantial interest in the geroscience and longevity research communities.
The Mitochondrial Theory of Aging
The relationship between mitochondria and aging has been discussed since Denham Harman first proposed the free radical theory of aging in 1956, later refined into the mitochondrial theory of aging. The core concept is straightforward: mitochondria are both the primary source and the primary target of reactive oxygen species within the cell. Over a lifetime, cumulative oxidative damage to mitochondrial DNA, proteins, and lipids progressively impairs mitochondrial function, creating a feed-forward cycle of increasing ROS production and decreasing ATP synthesis.
Modern geroscience has expanded this picture considerably. It's now understood that mitochondrial dysfunction contributes to aging not only through oxidative damage but also through altered signaling, impaired calcium handling, disrupted apoptosis regulation, and changes in the nuclear-mitochondrial communication that coordinates cellular metabolism. The concept of "mitochondrial dysfunction" as an aging hallmark encompasses all of these interconnected changes, not just free radical damage.
Cardiolipin sits at the center of many of these age-related changes. As noted earlier, cardiolipin content decreases 30-50% in multiple tissues of aged animals, and the remaining cardiolipin shows increased levels of peroxidation. These changes directly impair electron transport chain function, reduce ATP production, increase ROS generation, and sensitize cells to apoptotic death. The question that SS-31 research addresses is whether these changes are reversible, and the preclinical data strongly suggests they are.
Cardiac Aging: Reversing Diastolic Dysfunction
Diastolic dysfunction, the impaired relaxation and filling of the heart's ventricles during the resting phase of the cardiac cycle, is the most common age-related cardiac abnormality. It affects over 50% of individuals aged 70 and older and is the predominant cause of heart failure with preserved ejection fraction (HFpEF), a condition with no proven pharmacological therapy. The mitochondrial basis of diastolic dysfunction is well established: impaired ATP production reduces the energy available for active relaxation of the cardiac muscle, which, counterintuitively, requires more ATP than contraction.
A landmark study published in eLife by Chiao and colleagues (2020) demonstrated that 8 weeks of SS-31 treatment in old mice (24 months, equivalent to approximately 70 human years) substantially reversed diastolic dysfunction. Using echocardiography, pressure-volume loop analysis, and molecular assessments, the researchers showed that elamipretide treatment improved diastolic function to levels approaching those of young mice. The improvements correlated with restored mitochondrial function, normalized cardiolipin content, and reduced oxidative stress in cardiac tissue.
Perhaps most remarkably, the treatment was initiated in mice that were already old and already had established diastolic dysfunction. This wasn't a prevention study; it was a reversal study. The results demonstrated that age-related cardiac mitochondrial dysfunction is not a permanent condition but a potentially treatable state. This finding has profound implications for the management of diastolic heart failure in elderly humans, a condition that currently lacks effective pharmacotherapy.
Skeletal Muscle Aging: Restoring Exercise Capacity
Age-related skeletal muscle decline (sarcopenia) affects virtually all individuals to some degree, with significant impacts on mobility, independence, and quality of life. Mitochondrial dysfunction is a primary driver of sarcopenia: aged skeletal muscle shows reduced mitochondrial content, impaired oxidative phosphorylation, increased ROS production, and poor mitochondrial quality control (mitophagy).
Siegel and colleagues (2013) demonstrated that one hour of SS-31 treatment restored mitochondrial energetics in aged skeletal muscle. Specifically, SS-31 reversed the age-related decline in maximum mitochondrial ATP production (ATPmax) and improved the coupling efficiency of oxidative phosphorylation (P/O ratio) without increasing mitochondrial content. This is a critical distinction: the aged mitochondria were dysfunctional but not irreversibly damaged. By restoring their membrane integrity, SS-31 allowed existing mitochondria to function at near-youthful levels.
The functional consequences were significant. Treated aged mice showed increased treadmill endurance, greater fatigue resistance in isolated muscle preparations, and significantly greater gastrocnemius muscle mass compared to aged controls. The restoration of muscle function occurred without an increase in mitochondrial biogenesis markers, confirming that the mechanism was optimization of existing mitochondria rather than replacement with new ones.
Follow-up studies by Campbell and colleagues (2019) showed that improving mitochondrial function with SS-31 also reversed age-related redox stress and improved exercise tolerance in aged mice. The gastrocnemius muscle of treated mice demonstrated restored glutathione redox balance, normalized hydrogen peroxide emission rates, and improved calcium handling, all of which contributed to better contractile function.
These findings complement the benefits observed with other approaches to age-related decline. Growth hormone secretagogues like CJC-1295/Ipamorelin and Sermorelin address muscle decline through anabolic pathways, while BPC-157 and TB-500 support tissue repair mechanisms. SS-31's unique contribution is targeting the bioenergetic foundation that underlies muscle function, potentially working in conjunction with these other approaches. The Biohacking Hub covers strategies for combining various compounds to support optimal function.
Brain Aging: Neuroprotection and Cognitive Function
The brain consumes approximately 20% of the body's total energy despite representing only 2% of body mass, making neurons especially vulnerable to mitochondrial dysfunction. Age-related cognitive decline correlates with reduced cerebral mitochondrial function, and neurodegenerative diseases like Alzheimer's and Parkinson's involve pronounced mitochondrial pathology.
Preclinical studies have demonstrated neuroprotective effects of SS-31 in multiple contexts. In aged mice, SS-31 treatment improved synaptic mitochondrial function, reduced oxidative stress markers in brain tissue, and improved performance on memory and learning tasks. In models of focal cerebral ischemia (stroke), SS-31 reduced infarct volume and improved neurological outcomes. In a Parkinson's disease model using MPTP (a mitochondrial toxin), SS-31 protected dopaminergic neurons from degeneration.
A 2025 study by Tarantini and colleagues investigated SS-31's effects on age-related cerebral microvascular pathology, demonstrating that elamipretide treatment improved mitochondrial function in cerebrovascular endothelial cells, reduced cerebral microhemorrhages, and preserved blood-brain barrier integrity in aged mice. These findings highlight the vascular component of brain aging and suggest that SS-31's benefits extend beyond neuronal protection to encompass the cerebrovascular system that supports brain function.
The neuroprotective properties of SS-31 complement other peptides being studied for cognitive support. Semax, Selank, and Dihexa act through neurotrophic and nootropic mechanisms, while Pinealon targets peptide-mediated neuroprotection. SS-31's mitochondrial mechanism represents a distinct and complementary approach.
Mitochondrial Quality Control and SS-31
Beyond directly stabilizing cardiolipin, SS-31 appears to influence the quality control mechanisms that maintain a healthy mitochondrial population. These mechanisms include mitochondrial fission and fusion (the dynamic processes by which mitochondria divide and merge), mitophagy (the selective degradation of damaged mitochondria by autophagy), and mitochondrial biogenesis (the production of new mitochondria). Together, these processes constitute the "mitochondrial quality control network" that keeps the mitochondrial population functional.
In aged tissues, all three components of the quality control network become impaired. Mitochondrial dynamics shift toward excessive fission, producing small, fragmented mitochondria that function poorly. Mitophagy becomes less efficient, allowing damaged mitochondria to accumulate rather than being cleared. And mitochondrial biogenesis declines, reducing the rate at which new, healthy mitochondria are produced to replace damaged ones. The net result is a progressively deteriorating mitochondrial population.
SS-31 appears to improve mitochondrial quality control through indirect mechanisms. By stabilizing the inner membrane, SS-31 may restore the membrane conditions required for normal fission and fusion dynamics. The 2024 Barth syndrome study showed that SS-31 treatment normalized the expression of proteins involved in mitochondrial dynamics (including MFN1, MFN2, OPA1, and DRP1) and reactivated mitophagy pathways in tafazzin-deficient hearts. Similar findings in aged tissues suggest that many of the quality control defects attributed to aging are secondary consequences of cardiolipin deterioration rather than primary defects, and they can be reversed by restoring cardiolipin function.
This has important implications for understanding how SS-31 produces sustained benefits during long-term treatment. Rather than simply propping up dysfunctional mitochondria, SS-31 may create the conditions for the mitochondrial quality control network to restore a healthy mitochondrial population over time. This would explain the progressive improvement observed in the TAZPOWER extension: initial cardiolipin stabilization improves existing mitochondrial function (producing early benefits), while restored quality control gradually replaces the most damaged mitochondria with better-functioning ones (producing progressive long-term improvement).
Metabolic Implications of SS-31 in Aging
Age-related mitochondrial dysfunction has metabolic consequences that extend beyond ATP production. Mitochondria are central hubs for fatty acid oxidation, amino acid metabolism, ketone body production, and the citric acid cycle. When mitochondrial function declines, these metabolic pathways are impaired, contributing to the metabolic inflexibility and insulin resistance that characterize aging.
In aged mice, SS-31 treatment improved markers of metabolic health alongside its effects on mitochondrial function. Treated animals showed improved insulin sensitivity, reduced lipid accumulation in non-adipose tissues (a marker of metabolic dysfunction called ectopic lipid deposition), and better metabolic flexibility (the ability to switch between fat and carbohydrate oxidation based on substrate availability). These metabolic improvements occurred without changes in body weight or food intake, suggesting that they were driven by improved mitochondrial fat oxidation rather than altered energy balance.
The metabolic dimension of SS-31's effects connects it to the broader field of metabolic therapeutics. GLP-1 receptor agonists like semaglutide and tirzepatide improve metabolic health primarily through appetite regulation, weight loss, and direct effects on pancreatic beta cells. Tesofensine targets central appetite regulation. SS-31 approaches metabolism from the opposite direction: by improving the cellular machinery that processes nutrients into energy, it may enhance the metabolic benefits achieved through dietary and pharmacological interventions. The GLP-1 Weight Loss Overview covers metabolic approaches in detail.
Renal Aging: Preserving Kidney Function
Age-related decline in kidney function is nearly universal. Glomerular filtration rate (GFR) decreases by approximately 1 mL/min/year after age 40, and the prevalence of chronic kidney disease rises sharply in older populations. Mitochondrial dysfunction in renal tubular cells, which are highly dependent on oxidative phosphorylation for their reabsorptive functions, is a major driver of this decline.
Sweetwyne and colleagues (2017) published a study in Kidney International showing that SS-31 treatment improved glomerular architecture in aged mice. The treated animals showed less glomerulosclerosis, reduced podocyte foot process effacement, and better preservation of the glomerular filtration barrier compared to untreated aged controls. The improvements correlated with restored mitochondrial function in glomerular and tubular cells.
In models of acute kidney injury (AKI), SS-31 has shown protective effects through preservation of tubular cell mitochondrial function, reduction of oxidative stress, and attenuation of inflammatory responses. In a model of diabetic nephropathy, SS-31 protected against chronic mitochondrial damage and reduced renal fibrosis. A Phase 2a clinical trial also investigated elamipretide during stent revascularization in patients with atherosclerotic renal artery stenosis, demonstrating reduced post-procedural hypoxia and improved renal blood flow.
Ophthalmic Aging: Macular Degeneration
Age-related macular degeneration (AMD) is the leading cause of vision loss in individuals over 60, affecting more than 200 million people worldwide. The retinal pigment epithelium (RPE), which supports photoreceptor function, is among the most mitochondria-rich tissues in the body. With aging, RPE mitochondrial dysfunction leads to impaired clearance of photoreceptor outer segments, accumulation of lipofuscin and drusen, and progressive degeneration of the macula.
The ReCLAIM clinical trial program evaluated subcutaneous elamipretide in patients with dry AMD. The Phase 1 ReCLAIM studies (in both high-risk drusen and noncentral geographic atrophy cohorts) demonstrated safety and suggested possible improvements in low-luminance visual function after 24 weeks of daily elamipretide treatment.
The Phase 2 ReCLAIM-2 trial was a randomized, double-blind, placebo-controlled study evaluating elamipretide in patients with dry AMD and geographic atrophy. The primary endpoints of improvement in low-luminance best-corrected visual acuity and reduction in geographic atrophy area were not met. However, elamipretide was associated with a slowing of ellipsoid zone degradation, a structural endpoint that predicts progressive vision loss and AMD progression. This finding suggested that while elamipretide didn't produce immediate visual improvement, it may slow the structural deterioration that precedes vision loss.
Hearing, Balance, and Sensory Aging
Emerging research suggests that mitochondrial dysfunction also contributes to age-related sensory decline, including hearing loss (presbycusis) and vestibular dysfunction. The cochlear hair cells and stria vascularis cells that support hearing are metabolically active and mitochondria-dependent. Age-related damage to cochlear mitochondria contributes to the progressive hearing loss that affects approximately one-third of individuals over age 65.
While SS-31 has not been specifically tested in clinical trials for hearing loss, preclinical studies have shown that mitochondria-targeted antioxidants can protect against noise-induced and age-related cochlear damage. The demonstrated ability of SS-31 to reach multiple tissue types and its general mechanism of cardiolipin stabilization suggest potential applicability to auditory and vestibular aging, though this remains speculative pending dedicated research.
Skin and Connective Tissue Aging
Mitochondrial dysfunction in dermal fibroblasts and keratinocytes contributes to the visible signs of skin aging: wrinkles, loss of elasticity, thinning, and impaired wound healing. Dermal fibroblasts from older individuals show reduced mitochondrial function, increased ROS production, and decreased collagen synthesis compared to cells from younger individuals. The connection between mitochondrial energetics and collagen production is direct: collagen synthesis is one of the most ATP-intensive biosynthetic processes in the body, requiring substantial energy for amino acid hydroxylation, glycosylation, and triple helix formation.
While SS-31 is administered subcutaneously (and therefore passes through the skin), its primary distribution is systemic rather than local. For those specifically interested in skin health, topical peptides like GHK-Cu topical, SNAP-8, and Matrixyl address skin aging through direct local effects on collagen production, muscle relaxation, and extracellular matrix remodeling. SS-31's systemic effects on mitochondrial function may complement these topical approaches by improving the bioenergetic foundation that supports skin cell function throughout the body.
The Broader Anti-Aging Hypothesis
Taken together, the preclinical data across cardiac, skeletal muscle, neural, renal, and retinal tissues paints a consistent picture: age-related mitochondrial dysfunction is a treatable condition, and SS-31 can restore function in aged mitochondria without requiring mitochondrial replacement or biogenesis. This finding challenges the long-held assumption that mitochondrial aging is irreversible and opens the possibility of pharmacological interventions that could meaningfully slow or reverse aspects of biological aging.
The concept aligns with the broader geroscience hypothesis: that targeting the fundamental mechanisms of aging can simultaneously benefit multiple age-related diseases. Rather than treating heart failure, sarcopenia, cognitive decline, and kidney disease as separate conditions with separate treatments, a mitochondrial-targeted approach like SS-31 could address the common upstream mechanism that drives all of them.
Other compounds in the longevity peptide space, such as Epithalon (which targets telomere maintenance), FOXO4-DRI (which targets senescent cells), and Humanin (a mitochondria-derived peptide with cytoprotective properties), address different hallmarks of aging. The emerging picture suggests that optimal approaches to healthy aging may involve multiple interventions targeting different hallmarks simultaneously, with mitochondrial function as a foundation. Use the dosing calculator for personalized guidance on any of these compounds, and visit the Peptide Research Hub for detailed profiles of each.
Connecting Mitochondrial Dysfunction to Specific Aging Phenotypes
The research on SS-31 in aging models helps clarify the specific mechanisms by which mitochondrial dysfunction produces the phenotypic changes associated with aging. Rather than a vague "cellular energy decline," the data points to specific pathological cascades that can be interrupted by cardiolipin stabilization.
In the vascular system, age-related mitochondrial dysfunction in endothelial cells reduces nitric oxide bioavailability, increases vascular stiffness, and promotes chronic low-grade inflammation (often termed "inflammaging"). SS-31 treatment in aged mice improved endothelial function, reduced aortic stiffness, and decreased circulating inflammatory markers. These vascular effects may partially explain the cardiovascular protection observed in preclinical cardiac studies, as improved vascular function reduces the afterload on the heart and improves coronary blood flow.
In the immune system, mitochondrial dysfunction contributes to the chronic inflammatory state that characterizes aging. Damaged mitochondria release mitochondrial DNA (mtDNA) and other damage-associated molecular patterns (DAMPs) that activate the innate immune system through pattern recognition receptors. This mitochondria-driven inflammation has been linked to virtually every age-related chronic disease. By maintaining mitochondrial integrity, SS-31 may reduce the release of mitochondrial DAMPs and attenuate the inflammaging process.
In the stem cell compartment, mitochondrial dysfunction has been implicated in the age-related decline of tissue regenerative capacity. Stem cells from aged organisms show reduced mitochondrial function, and restoring mitochondrial health in stem cells can improve their self-renewal and differentiation capacity. While direct studies of SS-31 on stem cell function are limited, the peptide's ability to improve mitochondrial function in aged tissues suggests potential benefits for tissue regeneration and repair.
The convergence of these tissue-specific effects paints a picture of aging as fundamentally a mitochondrial problem, at least in part. The decline in ATP production, the increase in ROS, the release of mitochondrial DAMPs, the loss of membrane integrity - these are not isolated events but interconnected consequences of the same upstream cause: progressive cardiolipin deterioration in the inner mitochondrial membrane. SS-31 addresses this upstream cause, which is why it can produce beneficial effects across so many different tissues and organ systems simultaneously.
SS-31 in the Context of Geroscience
The geroscience hypothesis proposes that by targeting the fundamental biology of aging, we can simultaneously prevent or delay multiple age-related chronic diseases. This contrasts with the traditional disease-by-disease approach to medicine, where each condition is treated independently without addressing the shared underlying biology. SS-31 is arguably one of the best examples of a geroscience-aligned therapeutic, because its mechanism of action (cardiolipin stabilization) addresses a biological process (mitochondrial dysfunction) that is recognized as a fundamental hallmark of aging.
The National Institute on Aging (NIA) has embraced the geroscience framework and has called for clinical trials that test interventions against multiple aging outcomes simultaneously. SS-31 would be an excellent candidate for such trials, given its preclinical evidence of benefit across cardiac, muscle, renal, neural, and vascular aging phenotypes. The challenge lies in designing clinical trials that can capture these multi-organ benefits within realistic timeframes and budgets.
Other compounds that align with the geroscience approach include rapamycin analogs (targeting mTOR and cellular senescence), senolytics (clearing senescent cells), metformin (improving metabolic homeostasis), and the longevity peptides available through FormBlends. Epithalon targets telomere biology. FOXO4-DRI targets cellular senescence. GHK-Cu supports tissue repair and has shown anti-inflammatory effects. Together, these compounds represent a toolkit for addressing multiple hallmarks of aging, with SS-31 providing the mitochondrial foundation.
The Lifestyle Hub covers non-pharmaceutical approaches to supporting mitochondrial function and healthy aging, including exercise protocols, dietary strategies, and sleep optimization that complement peptide-based interventions.
Exercise Performance Research

Exercise capacity depends directly on mitochondrial function in skeletal and cardiac muscle. The ability of SS-31 to restore mitochondrial energetics has prompted research into its effects on exercise performance, with studies spanning preclinical aging models, primary mitochondrial myopathy, and healthy older adults.
Preclinical Exercise Data in Aged Animals
The most compelling preclinical exercise data comes from studies in aged mice. Siegel and colleagues demonstrated that SS-31 treatment reversed the age-related decline in ATPmax in gastrocnemius muscle, the primary calf muscle used in walking and running. Treated aged mice showed a 30% increase in ATPmax compared to untreated aged controls, bringing their mitochondrial ATP production back to approximately 85% of young healthy levels.
This bioenergetic improvement translated directly into functional gains. On a treadmill endurance test, SS-31-treated aged mice ran approximately 50% longer than untreated aged controls before reaching exhaustion. The gastrocnemius muscle was more fatigue-resistant in ex vivo stimulation experiments, maintaining force production over repeated contractions at levels comparable to young muscle. And the gastrocnemius muscle mass was significantly greater in treated aged mice, suggesting that SS-31 may help preserve lean muscle tissue during aging.
Campbell and colleagues (2019) extended these findings by showing that the exercise improvements were accompanied by restored redox homeostasis. In aged untreated muscle, hydrogen peroxide emission from mitochondria was elevated, the glutathione system was shifted toward the oxidized state, and calcium handling was impaired, all factors that contribute to muscle fatigue and weakness. SS-31 normalized all of these parameters, providing a mechanistic explanation for the functional improvements observed on exercise testing.
Clinical Trial: ATP Production in Older Adults
A randomized, double-blind, placebo-controlled clinical trial published in PLOS ONE by Ghonim and colleagues (2021) provided the first direct evidence that elamipretide can improve mitochondrial function in human skeletal muscle. The study enrolled older adults (ages 60-85) and measured in vivo mitochondrial ATP production in the first dorsal interosseous (FDI) muscle of the hand using 31-phosphorus magnetic resonance spectroscopy (31P-MRS), a non-invasive technique that quantifies the rate of ATP regeneration following exercise-induced ATP depletion.
Participants received a single 2-hour intravenous infusion of elamipretide or placebo. The results showed that a single dose of elamipretide significantly improved ATPmax in the FDI muscle compared to placebo. This was the first demonstration in humans that elamipretide could acutely improve mitochondrial ATP production in skeletal muscle, confirming the translational relevance of the preclinical findings.
The study had several strengths: it used a gold-standard measurement of in vivo mitochondrial function (31P-MRS), it employed a rigorous randomized double-blind design, and it focused on a directly relevant endpoint (ATP production in skeletal muscle). The limitation was its acute, single-dose design, which didn't address whether chronic treatment would produce sustained or progressive improvements in exercise capacity.
Primary Mitochondrial Myopathy Trials
Primary mitochondrial myopathy (PMM) refers to a group of genetic disorders caused by mutations in mitochondrial or nuclear DNA that directly impair oxidative phosphorylation. Patients with PMM suffer from profound exercise intolerance, muscle weakness, and fatigue, making them a natural population for studying mitochondrial-targeted therapies.
An initial randomized crossover trial evaluated subcutaneous elamipretide (40 mg daily for 5 days) in adults with genetically confirmed PMM. The primary endpoint was distance walked on the 6-minute walk test (6MWT). Elamipretide treatment increased 6MWT distance compared to placebo, with improvements emerging as early as Day 5 of treatment. The effect was particularly pronounced in patients with the most severe baseline exercise limitation.
The larger MMPOWER-3 trial was a Phase 3, randomized, double-blind, placebo-controlled study evaluating 24 weeks of daily subcutaneous elamipretide (40 mg) in 218 patients with PMM. Unfortunately, the trial did not meet its co-primary endpoints of change from baseline in 6MWT distance and the Primary Mitochondrial Myopathy Symptom Assessment (PMMSA) total fatigue score at Week 24. However, the trial confirmed the favorable safety profile of long-term elamipretide administration, and subgroup analyses suggested potential benefits in certain patient populations, particularly those with specific genetic subtypes of PMM.
The failure of MMPOWER-3 to meet its primary endpoints was a significant setback for the PMM indication, but it highlighted the challenges of studying heterogeneous genetic diseases with a single therapeutic approach. PMM encompasses dozens of different genetic mutations affecting different components of the mitochondrial machinery, and it's plausible that cardiolipin stabilization benefits some subtypes more than others.
Heart Failure and Exercise Intolerance
Exercise intolerance is a cardinal symptom of heart failure, driven by both central (cardiac output) and peripheral (skeletal muscle) factors. In dogs with experimentally induced heart failure, Sabbah and colleagues showed that elamipretide improved skeletal muscle mitochondrial function and reduced markers of oxidative stress, suggesting that the peptide addresses the peripheral component of exercise intolerance in addition to any central cardiac effects.
This dual mechanism (cardiac and skeletal muscle improvement) is particularly relevant for heart failure patients, in whom exercise intolerance persists even when cardiac function is optimized with standard therapies. The peripheral muscle mitochondrial dysfunction that develops during heart failure contributes to the persistent fatigue, dyspnea, and exercise limitation that dramatically impact quality of life.
Implications for Exercise and Aging
The exercise performance data for SS-31 has attracted interest beyond the disease context. The observation that acute elamipretide treatment can improve mitochondrial ATP production in healthy older adults raises questions about its potential as an adjunct to exercise training in aging populations. While exercise itself is the most potent stimulus for mitochondrial biogenesis and functional improvement in skeletal muscle, SS-31 might enhance the benefits of exercise by optimizing the function of existing mitochondria, allowing aged individuals to train more effectively and recover more completely.
This concept, sometimes called "exercise mimetics" or "exercise enhancers," has generated considerable discussion in the geroscience community. The distinction is important: SS-31 is not proposed as a replacement for exercise but as a potential complement that could lower the barrier to effective physical activity in individuals whose mitochondrial dysfunction limits their exercise capacity. For someone who can't walk far enough to generate a meaningful training stimulus, restoring even a portion of their mitochondrial capacity might enable them to exercise sufficiently to trigger adaptive remodeling.
Other peptides with relevance to physical performance include AOD-9604 (studied for body composition), MK-677 (a growth hormone secretagogue), and Tesamorelin (which increases growth hormone secretion). Each operates through a different mechanism, and the optimal approach to supporting physical performance likely involves addressing multiple physiological systems. MOTS-c, a mitochondria-derived peptide, also shows promise for exercise-related benefits through metabolic regulation. Those exploring these options can begin with a free assessment to identify their individual needs.
Mechanisms of Exercise Benefit: Detailed Pathway Analysis
The improvement in exercise performance with SS-31 involves multiple interconnected pathways, each stemming from the fundamental restoration of cardiolipin-dependent mitochondrial function. Understanding these pathways provides insight into why the exercise benefits are so consistent across different experimental models.
At the most basic level, exercise performance depends on the rate at which muscle cells can produce ATP. During high-intensity exercise, ATP turnover in skeletal muscle can increase 100-fold above resting levels. This demand can only be met by oxidative phosphorylation in mitochondria; glycolysis alone cannot sustain high-intensity effort for more than about 90 seconds. When mitochondrial function is impaired by age, disease, or genetic mutation, the maximum rate of ATP production decreases, and the threshold at which anaerobic metabolism must supplement aerobic metabolism (the anaerobic threshold) shifts downward. The result is earlier fatigue, greater lactate accumulation, and reduced exercise capacity.
SS-31 increases ATPmax by stabilizing the respiratory chain machinery, allowing more efficient electron transfer and proton pumping. In aged mice, this increase averages about 30% above untreated aged controls, recovering approximately 85% of youthful ATP production capacity. In older humans, a single dose improved ATPmax in the FDI muscle as measured by 31P-MRS, confirming that this mechanism operates in human tissue.
Beyond raw ATP production, SS-31 improves the coupling efficiency of oxidative phosphorylation. The P/O ratio (the number of ATP molecules produced per oxygen atom consumed) reflects how efficiently the proton gradient is converted to chemical energy. When the inner mitochondrial membrane leaks protons (a common consequence of cardiolipin damage), the P/O ratio drops, meaning more oxygen is consumed and more heat is generated for each ATP produced. SS-31 reduces proton leak by stabilizing membrane integrity, improving the P/O ratio and allowing more ATP production per unit of oxygen consumed. This translates directly into improved exercise efficiency, the ability to perform more work with less oxygen consumption.
SS-31 also reduces exercise-induced oxidative stress. During intense exercise, mitochondrial ROS production increases significantly, contributing to acute muscle fatigue and, over time, to exercise-induced muscle damage. By reducing electron leak at Complexes I and III, SS-31 decreases exercise-induced ROS production, potentially delaying the onset of oxidative fatigue and reducing post-exercise muscle damage. Campbell and colleagues confirmed this by showing that SS-31 normalized hydrogen peroxide emission rates and restored glutathione balance in aged mouse muscle.
Calcium handling is another pathway through which SS-31 may improve exercise performance. Mitochondria play a critical role in muscle calcium homeostasis, taking up calcium during contraction and releasing it during relaxation. This calcium cycling is an ATP-dependent process that can be impaired when mitochondrial function declines. In aged muscle, impaired mitochondrial calcium handling contributes to slower contraction-relaxation kinetics and reduced force production. SS-31 treatment improved calcium handling in aged muscle, supporting better contractile function.
Exercise Performance in Disease Populations
The exercise performance effects of SS-31 are particularly relevant in disease populations where exercise intolerance is a primary symptom. In primary mitochondrial myopathy, patients have genetic defects in the oxidative phosphorylation machinery itself, leading to profound exercise intolerance. The initial dose-escalation trial showed that just 5 days of elamipretide treatment improved 6-minute walk distance in PMM patients, a remarkably rapid effect consistent with SS-31's fast-acting mechanism.
In Barth syndrome, the 96.1-meter improvement in 6-minute walk distance over 168 weeks of treatment is clinically transformative. For patients who previously struggled to walk the length of a school hallway, an improvement of nearly 100 meters translates into meaningful gains in daily function, independence, and quality of life. The sustained nature of the improvement (continuing throughout the entire 168-week extension) suggests that elamipretide doesn't just provide a temporary boost but creates lasting improvements in muscle bioenergetics.
In heart failure, exercise intolerance is driven by both central (cardiac output) and peripheral (skeletal muscle) factors. Preclinical data from Sabbah's group showed that elamipretide improved skeletal muscle mitochondrial function in heart failure dogs independently of its cardiac effects, suggesting that the peptide could address the peripheral component of exercise intolerance even before cardiac function fully recovers. This dual mechanism is particularly attractive for heart failure management, where exercise intolerance persists despite optimal medical therapy in many patients.
The exercise data connects to the broader ecosystem of performance-enhancing peptides. While CJC-1295/Ipamorelin and GHRP-6 support exercise capacity through growth hormone-mediated anabolic effects, and BPC-157/TB-500 blend aids recovery from exercise-induced tissue damage, SS-31 uniquely targets the energy production machinery that fuels exercise performance at its most fundamental level.
Safety & Clinical Status

The safety profile of elamipretide has been characterized across more than a dozen clinical trials involving hundreds of patients, spanning acute intravenous administration and chronic subcutaneous dosing for periods up to 168 weeks. The overall profile supports good tolerability, with injection site reactions representing the most common and clinically significant adverse events.
Injection Site Reactions: The Primary Safety Consideration
Injection site reactions (ISRs) are the most frequently reported adverse events with subcutaneous elamipretide. Across clinical trials, ISRs have been reported in approximately 80% of patients receiving daily subcutaneous injections. These reactions typically include some combination of pain, erythema (redness), pruritus (itching), induration (hardening), swelling, and bruising at the injection site.
Despite the high incidence, several factors mitigate the clinical significance of ISRs. The vast majority (over 90%) are classified as mild in severity. They tend to appear early in treatment, with 67% of patients who experienced ISRs reporting them on the first day of administration. Over time, many patients experience a reduction in the frequency and severity of ISRs, suggesting partial tolerance development. No patients in the long-term TAZPOWER extension (168 weeks) discontinued treatment due to ISRs, indicating that patients learned to manage these reactions effectively.
A Phase 1 crossover study specifically investigated interventions to mitigate ISRs following subcutaneous elamipretide administration. The study evaluated pre-treatment with oral antihistamines, topical corticosteroids, and ice application, with results suggesting that oral antihistamines and topical corticosteroids could reduce the severity and duration of ISRs. These management strategies have been incorporated into the prescribing information for FORZINITY.
Systemic Adverse Events
Beyond injection site reactions, the systemic adverse event profile of elamipretide has been reassuringly clean across clinical trials. The most commonly reported systemic adverse events include headache, dizziness, nausea, abdominal discomfort, and fatigue. These events have generally been mild to moderate in severity and have occurred at rates only modestly higher than placebo in double-blind trials.
Across all clinical trials, there have been no clinically significant effects on vital signs (blood pressure, heart rate, respiratory rate, temperature), no consistent changes in laboratory parameters (hematology, clinical chemistry, urinalysis), and no clinically meaningful effects on electrocardiographic parameters (QT/QTc interval, heart rate, conduction intervals). This is particularly reassuring given that elamipretide targets a fundamental cellular process (mitochondrial function) and that off-target effects on cellular energetics could theoretically produce widespread physiological perturbations.
Rare instances of urticaria (hives) have been reported, suggesting the possibility of allergic-type reactions in susceptible individuals. However, these events have been uncommon and manageable with standard antihistamine therapy.
Long-Term Safety Data
The longest safety dataset for elamipretide comes from the TAZPOWER open-label extension, in which Barth syndrome patients received daily subcutaneous elamipretide for up to 168 weeks (approximately 3.2 years). This extended exposure dataset showed no accumulation of toxicity, no new safety signals emerging with prolonged treatment, and no clinically significant changes in laboratory values, vital signs, or cardiac monitoring over time.
Of the 10 patients who entered the open-label extension, 8 reached the Week 168 assessment. The two discontinuations were not related to adverse events from elamipretide but to other factors. This high retention rate in a pediatric/young adult population requiring daily injections speaks to both the tolerability of the treatment and the perceived benefit by patients and families.
Pharmacokinetic Considerations
The pharmacokinetic profile of elamipretide supports its safety. After subcutaneous injection, the peptide reaches peak plasma concentration within approximately 1 hour. The plasma half-life is relatively short (approximately 2-3 hours), meaning the compound is cleared rapidly from systemic circulation. However, the therapeutically relevant pharmacology depends on the peptide's residence time within mitochondrial membranes rather than its plasma concentration, so the short plasma half-life doesn't limit efficacy.
The rapid clearance from plasma reduces the risk of systemic accumulation during chronic dosing. At the approved dose of 40 mg subcutaneously once daily, steady-state pharmacokinetics show no evidence of accumulation over time. The peptide is metabolized primarily by peptidases into its component amino acids, which enter normal amino acid metabolism. There are no active metabolites requiring independent safety monitoring.
Drug Interactions
Elamipretide has a favorable drug interaction profile. As a small tetrapeptide metabolized by ubiquitous peptidases, it doesn't interact with cytochrome P450 enzymes, P-glycoprotein, or other common drug transporter systems. In clinical trials, elamipretide was administered alongside standard cardiovascular medications (ACE inhibitors, beta-blockers, diuretics, anticoagulants, statins) without evidence of pharmacokinetic or pharmacodynamic interactions.
This lack of drug interactions is clinically important because patients with conditions like heart failure, mitochondrial myopathy, and Barth syndrome typically take multiple medications. The ability to add elamipretide to existing regimens without dosing adjustments or interaction concerns simplifies clinical management.
Special Populations
The TAZPOWER trial enrolled patients as young as 12 years of age, providing some safety data in adolescents. The FORZINITY label specifies patients weighing at least 30 kg, with Stealth BioTherapeutics working to expand the indication to smaller children. The safety profile in the adolescent subgroup was consistent with that observed in adults.
Data in elderly patients comes primarily from the ReCLAIM AMD trials, which enrolled patients aged 55 and older. In this older population, elamipretide was well tolerated, with ISRs again being the most common adverse event. There were no age-specific safety concerns identified.
Pregnancy and lactation data are not available for elamipretide. Given the mechanism of action (cardiolipin stabilization in mitochondria) and the importance of mitochondrial function during embryonic development, use during pregnancy would require careful risk-benefit assessment. Animal reproductive toxicology studies have been conducted as part of the regulatory submission, but detailed results are not publicly available at this time.
Current Clinical Development Status
As of early 2026, the clinical development status of elamipretide spans several indications:
| Indication | Trial(s) | Status | Outcome |
|---|---|---|---|
| Barth Syndrome | TAZPOWER | FDA Approved (Sep 2025) | Approved as FORZINITY for muscle strength improvement |
| Primary Mitochondrial Myopathy | MMPOWER-3 | Completed | Did not meet primary endpoints; safety confirmed |
| Heart Failure (HFrEF) | PROGRESS-HF | Completed | Did not meet primary LVESV endpoint |
| Acute MI | EMBRACE-STEMI | Completed | Did not reduce infarct size; reduced HF incidence post-PCI |
| Dry AMD / Geographic Atrophy | ReCLAIM-2 | Completed | Did not meet primary visual endpoints; slowed ellipsoid zone loss |
| Renal Artery Stenosis | Phase 2a | Completed | Improved renal blood flow post-revascularization |
The pattern of clinical results, with consistent safety but mixed efficacy, reflects the challenges of translating a mitochondrial mechanism into clinical endpoints. The success in Barth syndrome, where the mitochondrial defect is the primary disease driver and the treatment duration was extended, suggests that elamipretide works best when mitochondrial dysfunction is the dominant pathological mechanism and when treatment is given long enough for mitochondrial restoration to produce measurable functional improvements.
For those exploring other peptides with established safety profiles, the Drug Comparison Hub provides side-by-side analyses of safety data across multiple compounds. The GLP-1 Weight Loss Overview covers the safety profiles of GLP-1 receptor agonists like semaglutide and tirzepatide, which have been through much larger clinical trial programs.
Comparison of Safety Across Clinical Indications
The safety profile of elamipretide has been remarkably consistent across different clinical populations, which is reassuring given the diversity of conditions studied. The following table summarizes the key safety findings across major clinical trials:
| Trial | Population | Route/Duration | Most Common AE | Serious AEs Related to Drug |
|---|---|---|---|---|
| Phase 1 (healthy volunteers) | Healthy adults | IV, single dose | Injection site pain (mild) | None |
| Early-phase HF | HFrEF (LVEF ≤35%) | IV, single dose | None significant over placebo | None |
| EMBRACE-STEMI | Acute MI patients | IV, 1-hour infusion | None significant over placebo | None related to drug |
| PROGRESS-HF | HFrEF (LVEF ≤40%) | SC, 28 days | Injection site reactions | None related to drug |
| TAZPOWER | Barth syndrome | SC, up to 168 weeks | Injection site reactions | None related to drug |
| MMPOWER-3 | Primary mitochondrial myopathy | SC, 24 weeks | Injection site reactions | None related to drug |
| ReCLAIM | Dry AMD, age 55+ | SC, 24 weeks | Injection site reactions | None related to drug |
The absence of serious drug-related adverse events across all trials is notable, particularly given that elamipretide targets a fundamental cellular process. The fact that improving mitochondrial function doesn't appear to cause off-target toxicity is consistent with the peptide's mechanism of action: rather than stimulating mitochondria to work harder (which could theoretically increase stress on already-damaged cells), SS-31 simply restores the structural conditions that allow mitochondria to function normally. It removes a brake rather than pressing the accelerator.
Practical Considerations for Clinical Use
For clinicians prescribing FORZINITY and for patients receiving treatment, several practical safety considerations are worth noting:
- Injection technique: Proper subcutaneous injection technique, including rotation of injection sites among the abdomen, thigh, and upper arm, helps minimize injection site reactions. Patients should be trained in self-injection during their initial clinical encounter.
- ISR management: Pre-treatment with a second-generation antihistamine (such as cetirizine or loratadine) 30-60 minutes before injection can reduce the severity of injection site reactions. Topical corticosteroid cream applied after injection may also help. Ice application before injection can reduce pain.
- Monitoring: No routine laboratory monitoring is required based on the clinical trial data. Standard clinical assessment is appropriate, with attention to injection sites for signs of infection or persistent inflammation.
- Storage: Elamipretide solution should be stored according to label instructions. Peptide stability should be verified before each administration.
- Drug interactions: No dose adjustments are needed for concurrent medications. Elamipretide can be safely co-administered with cardiovascular medications, immunosuppressants, and other standard therapies.
Understanding Injection Site Reactions: Mechanism and Management
The high incidence of injection site reactions (ISRs) with elamipretide warrants detailed discussion, as this is the primary tolerability concern for patients and prescribers. The mechanism of ISRs with elamipretide appears to be related to the peptide's physicochemical properties rather than a traditional immunological reaction.
SS-31 is a cationic peptide with membrane-active properties, designed to penetrate cell membranes rapidly. When injected subcutaneously, the high local concentration of the peptide at the injection site transiently interacts with the membranes of skin cells, mast cells, and sensory nerve endings in the dermis and subcutis. This interaction can trigger mast cell degranulation (releasing histamine and other mediators) and direct activation of nociceptive nerve fibers (causing pain and itching).
This mechanism explains several clinical observations. First, ISRs tend to be most prominent with the first injections and may diminish over time, consistent with mast cell depletion and desensitization of local sensory nerves. Second, pre-treatment with antihistamines (which block histamine-mediated effects) and topical corticosteroids (which reduce local inflammation) can mitigate ISR severity. Third, ice application before injection can reduce pain by slowing nerve conduction and vasoconstricting local vessels. Fourth, rotation of injection sites allows previously used sites to recover between injections.
For patients concerned about ISRs, it can be helpful to know that they are a local phenomenon with no systemic safety implications. They don't indicate an allergic reaction in the traditional sense, they don't worsen with continued treatment (and often improve), and they have not led to treatment discontinuation in any of the long-term clinical trials. The ISRs are a nuisance rather than a danger, and effective management strategies exist.
Theoretical Safety Considerations
Several theoretical safety concerns have been raised about long-term mitochondrial modulation with SS-31, none of which have materialized in clinical trials but which deserve discussion for completeness.
First, the question of whether improving mitochondrial function could promote cancer growth has been raised. Cancer cells often display altered mitochondrial function, with some relying more heavily on glycolysis (the Warburg effect) and others being critically dependent on oxidative phosphorylation. In principle, a drug that improves mitochondrial function could benefit cancer cells that rely on oxidative phosphorylation. However, the clinical trial data show no increased incidence of malignancy in elamipretide-treated patients, and the treatment durations (up to 168 weeks) are sufficient to detect any significant tumor-promoting effect. Additionally, SS-31's primary action of reducing ROS production could theoretically reduce DNA damage and mutation rates, providing a counterbalancing protective effect against cancer initiation.
Second, concerns about disrupting cellular quality control have been discussed. Mitophagy depends in part on the detection of damaged mitochondria through reduced membrane potential and externalized cardiolipin. If SS-31 masks the damage signals by stabilizing membranes and preventing cardiolipin externalization, it could theoretically impair the clearance of damaged mitochondria. However, the preclinical data actually shows the opposite: SS-31 treatment improves mitophagy efficiency rather than impairing it, likely because it restores the membrane conditions required for normal mitochondrial dynamics and quality control signaling.
Third, the question of whether chronic mitochondrial stimulation could accelerate telomere shortening or cellular senescence has been considered. This concern is based on the (debated) concept that increased metabolic activity accelerates cellular aging. Again, the evidence doesn't support this concern: SS-31 reduces ROS production (which drives telomere damage) and doesn't increase the total metabolic rate (it improves efficiency rather than throughput). These effects would, if anything, be expected to slow cellular aging rather than accelerate it.
Regulatory and Commercial Outlook
The accelerated approval of FORZINITY for Barth syndrome is contingent upon confirmatory trials demonstrating clinical benefit. Stealth BioTherapeutics is working with the FDA to design these confirmatory studies, which may include broader clinical endpoints beyond muscle strength.
The commercial viability of FORZINITY for Barth syndrome is constrained by the ultra-rare nature of the disease (approximately 150 patients in the United States). However, the approval establishes the regulatory precedent for mitochondrial-targeted therapeutics and may support future development of elamipretide or next-generation Szeto-Schiller peptides for more prevalent conditions.
Stealth BioTherapeutics continues to explore additional indications and has expressed interest in the aging-related conditions where preclinical data has been most promising. The challenge will be designing clinical trials that can capture the benefits of mitochondrial restoration in conditions where the endpoints are measured in years rather than weeks.
SS-31 in Neurological and Neurodegenerative Disease Research
The brain consumes roughly 20% of the body's total oxygen and energy despite representing only 2% of body mass. This extraordinary metabolic demand makes neurons uniquely vulnerable to mitochondrial dysfunction, and it's no surprise that mitochondrial deterioration is a shared feature of nearly every neurodegenerative disease. SS-31's ability to restore mitochondrial function at the inner membrane level makes it a compelling candidate for neurological applications, and the preclinical data in this area has been consistently encouraging.
Alzheimer's Disease and Amyloid-Beta Toxicity
In Alzheimer's disease (AD), mitochondrial dysfunction is now recognized as an early event that precedes amyloid plaque formation and tau tangle accumulation. The "mitochondrial cascade hypothesis" proposes that age-related decline in mitochondrial function triggers the amyloid processing pathway, rather than the other way around. This hypothesis reframes mitochondrial protection as a potential upstream intervention rather than a downstream supportive measure.
SS-31 has been tested extensively in transgenic mouse models of Alzheimer's disease. In the 3xTg-AD model, which develops both amyloid and tau pathology, chronic SS-31 administration reduced amyloid-beta production by approximately 40%, decreased tau phosphorylation, and improved performance on spatial memory tasks. The mechanism appears to involve restoration of normal mitochondrial calcium handling, which when disrupted, activates the beta-secretase enzyme (BACE1) that cleaves amyloid precursor protein into toxic amyloid-beta fragments.
The synaptic effects of SS-31 are particularly relevant to Alzheimer's. Synaptic mitochondria, which provide the ATP needed for neurotransmitter release and recycling, are more vulnerable to age-related damage than non-synaptic mitochondria. SS-31 preferentially improves the function of these synaptic mitochondria, restoring ATP production at nerve terminals and improving synaptic transmission. In AD model mice, this translated to measurably improved long-term potentiation (LTP), the cellular mechanism underlying learning and memory.
Parkinson's Disease and Dopaminergic Neuron Protection
Parkinson's disease (PD) has one of the strongest connections to mitochondrial dysfunction of any neurodegenerative condition. Mutations in genes encoding mitochondrial proteins (PINK1, Parkin, DJ-1) cause familial forms of the disease, and complex I deficiency in the mitochondrial electron transport chain is consistently observed in sporadic PD. The dopaminergic neurons of the substantia nigra, which are selectively lost in PD, have particularly high energy demands due to their extensive axonal arbors and pacemaker-like firing patterns.
In the MPTP mouse model of Parkinson's disease, SS-31 provided substantial neuroprotection. Pre-treatment with SS-31 reduced dopaminergic neuron loss by 60-70% and preserved striatal dopamine levels, which are the primary determinant of motor function. The protection was dose-dependent, and even post-treatment (beginning SS-31 after MPTP exposure) provided partial neuroprotection, suggesting a therapeutic window beyond just prophylaxis.
The 6-OHDA rat model, which targets dopaminergic neurons through oxidative stress, showed similar protective effects. SS-31-treated animals maintained better motor performance on rotational behavior tests and showed less tyrosine hydroxylase-positive neuron loss in the substantia nigra. These results across different PD models, each using a different mechanism of neurotoxicity, strengthen the case that SS-31's neuroprotection is mediated through a general mitochondrial mechanism rather than blocking a specific toxin.
Traumatic Brain Injury and Acute Neurological Insults
Traumatic brain injury (TBI) triggers a cascade of secondary injury events in which mitochondrial dysfunction plays a central role. The initial mechanical trauma disrupts mitochondrial membranes, leading to calcium overload, excessive ROS production, and energy failure. This secondary mitochondrial crisis can expand the zone of injury far beyond the initial impact site and continues for days to weeks after the initial trauma.
SS-31 administration within hours of experimental TBI in rodents has shown consistent neuroprotective effects. Treated animals showed smaller lesion volumes, reduced brain edema, lower levels of neuroinflammatory markers, and improved performance on cognitive and motor tests. The optimal treatment window appeared to be within 6 hours of injury, though some benefit was observed even when treatment was delayed by 12-24 hours.
For stroke (ischemic brain injury), the data is equally promising. SS-31 reduced infarct volume by 40-55% in middle cerebral artery occlusion models when administered before or shortly after the ischemic event. The mechanism involves preservation of mitochondrial membrane integrity during reperfusion, which prevents the burst of ROS production that typically occurs when blood flow is restored to ischemic tissue. This reperfusion injury is a major contributor to the final infarct size and is a logical target for mitochondrial-targeted therapy.
Retinal and Optic Nerve Degeneration
The retina and optic nerve are essentially extensions of the central nervous system, and their mitochondrial vulnerability mirrors that of the brain. Retinal ganglion cells, photoreceptors, and the retinal pigment epithelium all depend heavily on oxidative phosphorylation, making them susceptible to mitochondrial dysfunction with aging and disease.
SS-31 has shown protective effects in multiple models of retinal degeneration. In light-induced retinal damage models, SS-31 preserved photoreceptor structure and function as measured by electroretinography. In models of diabetic retinopathy, it reduced retinal vascular leakage and preserved visual acuity measures. And in glaucoma models, SS-31 protected retinal ganglion cells from pressure-induced death, maintaining axon integrity in the optic nerve.
The optic neuropathy application is especially relevant because Leber hereditary optic neuropathy (LHON), a mitochondrial genetic disease that causes bilateral vision loss in young adults, currently has very limited treatment options. SS-31's ability to improve electron transport chain function independent of specific genetic defects makes it a rational candidate for LHON and other mitochondrial optic neuropathies. While no clinical trials have been completed for these indications, the preclinical rationale is strong.
Connections to Other Neuroprotective Peptides
SS-31's neurological applications intersect with several other peptides that target brain health through complementary mechanisms. Semax enhances BDNF expression and provides neuroprotection through neurotrophic pathways. Selank modulates anxiety and cognitive function through GABAergic mechanisms. Dihexa promotes synaptic connectivity through hepatocyte growth factor signaling. And Pinealon targets central nervous system peptide regulation. Each approaches neuroprotection from a different angle, and combinations targeting multiple pathways simultaneously represent a growing area of interest in longevity research.
The mitochondria-focused approach of SS-31 also connects with other mitochondrial peptides. Humanin and MOTS-c are mitochondria-derived peptides that protect against cellular stress through signaling pathways distinct from SS-31's direct cardiolipin interaction. NAD+ supplementation supports mitochondrial function through the electron carrier pathway. Together, these approaches offer multiple entry points for addressing the mitochondrial dysfunction that underlies aging and neurodegenerative disease. The biohacking hub provides a broader view of these interconnected strategies, and the free assessment can help identify which approaches align with your specific health priorities.
SS-31 and Kidney Disease: Renal Mitochondrial Protection
The kidneys are second only to the heart in mitochondrial density, and renal tubular cells rely almost entirely on oxidative phosphorylation to power the active transport processes that filter and reabsorb nearly 180 liters of fluid per day. This extreme energy dependence makes the kidneys exquisitely vulnerable to mitochondrial dysfunction, which is why acute kidney injury (AKI) and chronic kidney disease (CKD) are increasingly recognized as mitochondrial diseases at their core.
SS-31 has shown protective effects across multiple models of kidney injury. In ischemia-reperfusion injury, which mimics the kidney damage that occurs during major surgery or transplantation, SS-31 administered before or shortly after the ischemic event reduced tubular cell death by 50-70%, preserved GFR (glomerular filtration rate), and accelerated recovery of renal function. The mechanism aligns with what's seen in cardiac ischemia-reperfusion: SS-31 maintains cardiolipin integrity during the ischemic period and prevents the burst of reactive oxygen species that typically occurs when blood flow returns.
In diabetic kidney disease, the most common cause of end-stage renal disease worldwide, SS-31 has demonstrated benefits in preclinical models that go beyond simple protection. In the db/db diabetic mouse model, chronic SS-31 administration reduced albuminuria (protein in the urine, a marker of kidney damage), preserved podocyte structure, and maintained normal mitochondrial morphology in renal tubular cells. The podocyte protection is particularly relevant, because podocyte loss is an irreversible event that directly drives glomerulosclerosis and progressive kidney function decline.
Cisplatin nephrotoxicity, a common and dose-limiting side effect of one of oncology's most widely used chemotherapy agents, presents another application where SS-31's protective profile is relevant. Cisplatin damages renal tubular cells primarily through mitochondrial injury, triggering ROS production, DNA damage, and apoptosis. In mouse models, SS-31 co-administration with cisplatin preserved kidney function and reduced histological evidence of tubular damage without compromising the anti-tumor efficacy of the chemotherapy. If these results translate to human patients, SS-31 could allow oncologists to use higher and more effective cisplatin doses without the kidney toxicity that currently limits treatment intensity.
The aging kidney deserves special mention. Age-related decline in kidney function affects virtually everyone, with estimated GFR declining approximately 1 mL/min/year after age 40. This decline is strongly correlated with mitochondrial dysfunction in renal tubular cells, including the same patterns of cardiolipin oxidation, electron transport chain impairment, and increased ROS production that SS-31 targets. In aged rats, SS-31 treatment restored renal mitochondrial function and improved age-related proteinuria, suggesting that the age-related decline in kidney function may be partially reversible through mitochondrial restoration.
The kidney also connects to several metabolic pathways affected by peptides in other classes. GLP-1 receptor agonists like semaglutide have demonstrated independent kidney-protective effects, and the combination of GLP-1-mediated hemodynamic and metabolic benefits with SS-31's direct mitochondrial protection represents an intriguing multi-target approach to renal preservation. BPC-157 has shown renal protective effects in some animal models through its cytoprotective and anti-inflammatory actions. The convergence of multiple peptide approaches on kidney health reflects the growing recognition that renal disease requires multi-pathway intervention rather than single-target therapy. The comparison hub provides detailed analyses of how different peptide compounds overlap and complement each other across organ systems.
Practical Considerations for Peptide Storage and Stability
For researchers and clinicians working with SS-31, proper handling is essential to maintaining the compound's biological activity. The lyophilized form is relatively stable and can be stored at -20 degrees Celsius for extended periods without significant degradation. However, once reconstituted in aqueous solution, SS-31 begins to oxidize, particularly at the dimethyltyrosine (Dmt) residue, which is critical for its cardiolipin binding activity. Reconstituted solutions should be stored at 2-8 degrees Celsius and used within 7 days, or aliquoted and frozen at -80 degrees Celsius for longer storage.
The choice of reconstitution vehicle matters for stability. Bacteriostatic water with 0.9% benzyl alcohol provides adequate preservation against microbial contamination for multi-dose use, but the preservative can potentially interact with the peptide over extended storage periods. Sterile water is preferred for immediate single-use preparations. Some researchers report improved stability when reconstituting in phosphate-buffered saline at pH 7.4, which more closely matches physiological conditions and may slow oxidative degradation.
Light exposure accelerates SS-31 degradation through photo-oxidation of the Dmt residue. Amber vials or aluminum foil wrapping should be used for both storage and during administration. Temperature cycling, repeated freezing and thawing, also compromises peptide integrity. Each freeze-thaw cycle can reduce potency by 5-10%, so preparing single-use aliquots from the initial reconstitution is strongly recommended for research applications where consistent dosing is critical.
These practical considerations apply broadly to most research peptides, including compounds like Epithalon, FOXO4-DRI, and GHK-Cu, all of which require similar attention to storage conditions to maintain their bioactivity. The science page provides additional guidance on peptide handling and quality verification.
Third-party purity testing through high-performance liquid chromatography (HPLC) and mass spectrometry is strongly recommended for any SS-31 obtained outside of clinical trial settings. Impurities in synthetic peptides can include truncated sequences, racemized amino acids, and residual coupling reagents, any of which can reduce efficacy or introduce confounding variables in research applications. A certificate of analysis showing greater than 95% purity by HPLC, with mass spectrometry confirmation of the correct molecular weight (640.8 Da for SS-31 free base), provides reasonable assurance of product identity and quality. Reputable suppliers like FormBlends provide third-party analytical documentation with each batch, giving researchers confidence in the consistency and identity of the material they are working with throughout their studies.
SS-31 and Exercise Performance: Mitochondrial Enhancement in Athletic and Age-Related Functional Decline
One of the most intriguing applications of SS-31 research lies at the intersection of mitochondrial biology and exercise physiology. Skeletal muscle is among the most mitochondria-dense tissues in the body, with mitochondria comprising 5-12% of total muscle fiber volume in trained individuals. The capacity for sustained physical work depends almost entirely on mitochondrial oxidative phosphorylation, and the decline in exercise capacity that accompanies aging correlates closely with deteriorating mitochondrial function in skeletal muscle. This relationship has prompted researchers to investigate whether SS-31's mitochondrial-targeting properties could improve exercise performance, accelerate recovery, or reverse the functional decline associated with aging muscle.
Preclinical studies in aged mice have provided compelling evidence. When 26-month-old mice (roughly equivalent to 70-year-old humans in terms of physiological aging) received SS-31 treatment for 8 weeks, they showed significant improvements in treadmill endurance, grip strength, and voluntary wheel running activity compared to age-matched controls receiving vehicle treatment. Critically, muscle biopsy analysis revealed that SS-31 treatment was associated with improved mitochondrial cristae structure, enhanced respiratory chain complex activity, and reduced mitochondrial hydrogen peroxide emission. These structural and functional improvements in muscle mitochondria correlated directly with the observed gains in physical performance.
The mechanism by which SS-31 improves exercise capacity appears to involve both acute and chronic effects. Acutely, SS-31's stabilization of cardiolipin in the inner mitochondrial membrane optimizes electron transport chain efficiency, reducing electron leak and increasing the ratio of ATP production to oxygen consumption. This means that for any given level of physical work, treated muscle generates more usable energy with less oxidative waste. Chronically, the reduction in mitochondrial reactive oxygen species production appears to create a more favorable environment for mitochondrial biogenesis and quality control through mitophagy, the selective removal of damaged mitochondria. Over weeks of treatment, the net effect is a shift toward a healthier, more functional mitochondrial population within muscle fibers.
The implications extend beyond athletic performance to the clinical problem of sarcopenia, the age-related loss of muscle mass and function that affects approximately 10-15% of adults over age 65 and up to 50% of those over age 80. Sarcopenia is a leading cause of falls, disability, loss of independence, and premature mortality in older adults, yet effective pharmacological treatments remain elusive. Current management relies primarily on resistance exercise and protein supplementation, which are helpful but insufficient for many elderly patients. SS-31's ability to improve mitochondrial function in aged muscle, even without concurrent exercise training, suggests it could serve as a pharmacological adjunct to exercise-based interventions for sarcopenia management.
For athletes and fitness-focused individuals, the research on SS-31 and exercise recovery is also noteworthy. Intense exercise generates substantial mitochondrial oxidative stress, which contributes to delayed onset muscle soreness, temporary performance decrements, and the inflammatory response that, while necessary for adaptation, can become counterproductive when excessive or prolonged. By reducing mitochondrial ROS production during and after exercise, SS-31 could theoretically accelerate recovery between training sessions without blunting the adaptive signaling that drives training adaptations. This distinction is important because indiscriminate antioxidant supplementation (such as high-dose vitamin C or E) has been shown in some studies to impair exercise adaptation by suppressing the redox-sensitive signaling pathways that trigger mitochondrial biogenesis. SS-31's targeted action at the inner mitochondrial membrane may allow it to reduce excessive oxidative damage while preserving the adaptive signaling that occurs in other cellular compartments.
Current research interest in SS-31 for exercise applications is growing, with several academic groups conducting or planning human studies. Researchers interested in mitochondrial support through SS-31 can explore the available research-grade material, and complementary approaches to mitochondrial health include the mitochondrial-derived peptide MOTS-c, which enhances exercise capacity through AMPK activation and improved glucose metabolism. The two peptides target different aspects of mitochondrial function and may provide complementary benefits when used in combination.
Frequently Asked Questions
What is SS-31 (elamipretide) and how does it work?
SS-31, also known as elamipretide, is a synthetic tetrapeptide with the amino acid sequence D-Arg-Dmt-Lys-Phe-NH2. It belongs to the Szeto-Schiller family of cell-permeable peptides designed to target mitochondria. The peptide crosses cell membranes rapidly and accumulates in mitochondria at concentrations 1,000 to 5,000 times higher than in the cytoplasm. Once inside mitochondria, SS-31 binds selectively to cardiolipin, a phospholipid found almost exclusively in the inner mitochondrial membrane. This binding stabilizes the electron transport chain complexes, reduces reactive oxygen species production, and improves ATP synthesis. Unlike earlier mitochondrial-targeting strategies, SS-31 doesn't depend on mitochondrial membrane potential for uptake, meaning it can reach damaged mitochondria that other targeting approaches cannot access.
What is cardiolipin and why is it important for mitochondrial function?
Cardiolipin is a unique dimeric phospholipid found almost exclusively in the inner mitochondrial membrane, where it constitutes roughly 20% of total lipid content. Its distinctive structure, with four fatty acyl chains and two phosphate headgroups, allows it to form the curved membrane structures (cristae) that increase the surface area for oxidative phosphorylation. Cardiolipin serves as an essential structural component and functional co-factor for all five electron transport chain complexes, the adenine nucleotide translocator, and cytochrome c. When cardiolipin becomes oxidized or depleted, whether from genetic mutations, ischemic damage, or aging, mitochondrial function deteriorates rapidly. ATP production falls, reactive oxygen species increase, and cells become more susceptible to death. SS-31 works by protecting and stabilizing cardiolipin, maintaining the optimal membrane environment for energy production.
Has elamipretide been approved by the FDA?
Yes. On September 19, 2025, the FDA granted accelerated approval to FORZINITY (elamipretide) injection for the improvement of muscle strength in adult and pediatric patients with Barth syndrome weighing at least 30 kilograms. This made FORZINITY the first FDA-approved treatment for Barth syndrome and the first approved mitochondria-targeted therapeutic. The approval was based on knee extensor muscle strength data from the open-label extension of the TAZPOWER clinical trial. Because this was an accelerated approval based on a surrogate endpoint (muscle strength), continued approval is contingent upon verification of clinical benefit in confirmatory trials. The approved dose is 40 mg administered subcutaneously once daily.
Can SS-31 treat heart failure?
SS-31 has been studied in heart failure but is not currently approved for this indication. An early-phase trial showed that intravenous elamipretide produced acute improvements in left ventricular volumes in HFrEF patients, with a significant reduction in end-diastolic volume of 18 mL and end-systolic volume of 14 mL in the highest dose group. However, the PROGRESS-HF trial (28 days of subcutaneous dosing) did not meet its primary endpoint of improved left ventricular end-systolic volume. The EMBRACE-STEMI trial in acute myocardial infarction also missed its primary infarct size endpoint, though it did show reduced incidence of heart failure within 24 hours of PCI. Preclinical data remains strong, with elamipretide restoring mitochondrial function and improving cardiac performance in animal models of heart failure.
What are the side effects of elamipretide?
The most common side effects of elamipretide are injection site reactions, reported in approximately 80% of patients receiving daily subcutaneous injections. These reactions include pain, redness, itching, swelling, hardening, and bruising at the injection site. The vast majority are mild in severity, and many patients experience a reduction in these reactions over time. Oral antihistamines and topical corticosteroids can help manage injection site symptoms. Other less common adverse events include headache, dizziness, nausea, abdominal discomfort, and fatigue, all generally mild to moderate. Long-term safety data from the 168-week TAZPOWER extension showed no accumulation of toxicity, no new safety signals, and no clinically significant changes in laboratory values or cardiac monitoring.
Is SS-31 an anti-aging compound?
SS-31 has shown remarkable anti-aging effects in preclinical studies, though it is not approved or marketed as an anti-aging therapy. In aged mice, SS-31 reversed age-related diastolic cardiac dysfunction, restored skeletal muscle mitochondrial function and exercise capacity, improved renal glomerular architecture, and protected cerebrovascular function. A clinical trial in older adults demonstrated that a single dose of elamipretide improved skeletal muscle ATP production measured by magnetic resonance spectroscopy. The consistent finding across these studies is that age-related mitochondrial dysfunction is not permanent but can be pharmacologically reversed by stabilizing cardiolipin on the inner mitochondrial membrane. However, large-scale clinical trials specifically evaluating anti-aging endpoints have not been conducted.
What is Barth syndrome and how does SS-31 help?
Barth syndrome is a rare, X-linked genetic disorder caused by mutations in the TAFAZZIN gene, which encodes an enzyme critical for cardiolipin remodeling. Without functional tafazzin, cardiolipin levels drop and abnormal cardiolipin species accumulate, leading to mitochondrial dysfunction throughout the body. Patients develop cardiomyopathy, skeletal muscle weakness, neutropenia, and growth retardation, often beginning in infancy. SS-31 helps by binding to the remaining cardiolipin in tafazzin-deficient mitochondria, partially compensating for the structural and functional deficits. In the TAZPOWER trial, long-term elamipretide treatment improved 6-minute walk distance by 96 meters, enhanced cardiac function, reduced fatigue, and increased muscle strength over 168 weeks. The FDA approved elamipretide (as FORZINITY) for Barth syndrome in September 2025.
How is elamipretide different from other mitochondrial supplements like CoQ10 or NAD+?
Elamipretide targets mitochondria through a fundamentally different mechanism than supplements like CoQ10 or NAD+. While CoQ10 serves as an electron carrier in the transport chain and NAD+ is a cofactor for mitochondrial enzymes (particularly in the TCA cycle and electron transport), elamipretide works at the structural level by stabilizing cardiolipin, the phospholipid that organizes and supports all of the respiratory chain machinery. This structural stabilization improves the efficiency of the entire electron transport chain rather than supplementing one component. Additionally, elamipretide concentrates in mitochondria at therapeutic levels through its unique cell-penetrating design, while oral CoQ10 and NAD+ supplements face absorption and bioavailability challenges that limit their mitochondrial delivery. These approaches are potentially complementary rather than competitive.
What is the dosing protocol for SS-31?
The FDA-approved dose of FORZINITY (elamipretide) for Barth syndrome is 40 mg administered subcutaneously once daily. This dose was established through the TAZPOWER clinical trial program and is the dose used in most subcutaneous clinical trials across indications. For intravenous administration (used in acute settings like the EMBRACE-STEMI trial), doses have ranged from 0.005 to 0.25 mg/kg/hr. The subcutaneous injection should be rotated among different injection sites (abdomen, thigh, upper arm) to minimize injection site reactions. Pre-treatment with oral antihistamines or application of topical corticosteroids can help manage injection site reactions. The peptide reaches peak plasma concentration within approximately 1 hour of subcutaneous injection, with a plasma half-life of 2-3 hours, though its therapeutic effect depends on residence time within mitochondrial membranes.
Can SS-31 improve exercise performance in healthy individuals?
There is limited clinical data on elamipretide's effects on exercise performance in healthy individuals. The available evidence comes primarily from a trial in older adults, where a single dose improved skeletal muscle ATP production measured by phosphorus magnetic resonance spectroscopy. In aged mice, SS-31 increased treadmill endurance by approximately 50% and improved muscle fatigue resistance. These results suggest potential benefit for individuals with age-related mitochondrial decline, but they don't necessarily apply to young, healthy individuals whose mitochondria are already functioning optimally. SS-31 appears to work by restoring dysfunctional mitochondria rather than enhancing already-healthy ones, meaning the benefit may be proportional to the degree of pre-existing mitochondrial impairment. Clinical trials in healthy populations specifically targeting exercise performance have not been conducted.
What other conditions is elamipretide being studied for?
Beyond its approved indication in Barth syndrome, elamipretide has been or is being studied in several conditions where mitochondrial dysfunction plays a role. These include primary mitochondrial myopathy (the MMPOWER-3 trial), heart failure with reduced ejection fraction (PROGRESS-HF), acute myocardial infarction (EMBRACE-STEMI), dry age-related macular degeneration and geographic atrophy (the ReCLAIM program), and atherosclerotic renal artery stenosis. Preclinical research has also explored the peptide in models of diabetic nephropathy, acute kidney injury, Parkinson's disease, stroke, Friedreich's ataxia, and general aging. The breadth of these investigations reflects the fundamental role of mitochondrial function in health and disease across every organ system.
How does SS-31 compare to other longevity peptides?
SS-31 occupies a unique position among longevity peptides by targeting the mitochondrial membrane directly. While Epithalon targets telomere maintenance through telomerase activation, FOXO4-DRI addresses cellular senescence, and Humanin acts as a mitochondria-derived signaling peptide, SS-31 works at the structural level of the organelle itself, stabilizing the lipid environment that supports energy production. MOTS-c, another mitochondria-derived peptide, regulates metabolic homeostasis through AMPK activation and nuclear gene regulation, representing a signaling rather than structural approach to mitochondrial health. NAD+ precursors support mitochondrial enzyme function. Each targets a different aspect of cellular aging, and they may be complementary. SS-31 is the most clinically advanced of these compounds, with FDA approval and extensive human safety data, making it a foundational element in mitochondrial-targeted longevity strategies.
References
- Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. British Journal of Pharmacology. 2014;171(8):2029-2050. DOI: 10.1111/bph.12461.
- Birk AV, Liu S, Soong Y, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. Journal of the American Society of Nephrology. 2013;24(8):1250-1261. DOI: 10.1681/ASN.2012121216.
- Zhao K, Zhao GM, Wu D, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. Journal of Biological Chemistry. 2004;279(33):34682-34690. DOI: 10.1074/jbc.M402999200.
- Pharaoh G, Kamat V, Kannan S, et al. Mitochondrial protein interaction field of SS-31. Proceedings of the National Academy of Sciences. 2020;117(26):15363-15373. DOI: 10.1073/pnas.2002250117.
- Birk AV, Chao WM, Bracken C, et al. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. British Journal of Pharmacology. 2014;171(8):2017-2028. DOI: 10.1111/bph.12468.
- Mitchell W, Ng EA, Taber JD, et al. The mitochondria-targeted peptide SS-31 binds lipid bilayers and modulates surface electrostatics as a key component of its mechanism of action. Journal of Biological Chemistry. 2020;295(21):7452-7469. DOI: 10.1074/jbc.RA119.012094.
- Sabbah HN, Gupta RC, Kohli S, et al. Chronic therapy with elamipretide (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circulation: Heart Failure. 2016;9(2):e002206. DOI: 10.1161/CIRCHEARTFAILURE.115.002206.
- Daubert MA, Yow E, Duber G, et al. Novel mitochondria-targeting peptide in heart failure treatment: a randomized, placebo-controlled trial of elamipretide. Circulation: Heart Failure. 2017;10(12):e004389. DOI: 10.1161/CIRCHEARTFAILURE.117.004389.
- Butler J, Khan MS, Anker SD, et al. Effects of elamipretide on left ventricular function in patients with heart failure with reduced ejection fraction: the PROGRESS-HF Phase 2 trial. Journal of Cardiac Failure. 2020;26(5):429-437. DOI: 10.1016/j.cardfail.2020.02.001.
- Thompson R, Kang J, Yin B, et al. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in Barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genetics in Medicine. 2021;23(3):471-478. DOI: 10.1038/s41436-020-01006-8.
- Hornby B, Thompson WR, Alber J, et al. Long-term efficacy and safety of elamipretide in patients with Barth syndrome: 168-week open-label extension results of TAZPOWER. Genetics in Medicine. 2024;26(7):101131. DOI: 10.1016/j.gim.2024.101131.
- Thompson WR, Hornby B, Manuel R, et al. Natural history comparison study to assess the efficacy of elamipretide in patients with Barth syndrome. Orphanet Journal of Rare Diseases. 2022;17(1):336. DOI: 10.1186/s13023-022-02469-5.
- Chatfield KC, Sparagna GC, Chau S, et al. SS-31 treatment ameliorates cardiac mitochondrial morphology and defective mitophagy in a murine model of Barth syndrome. Scientific Reports. 2024;14:13804. DOI: 10.1038/s41598-024-64368-y.
- Chiao YA, Zhang H, Sweetwyne M, et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. eLife. 2020;9:e55513. DOI: 10.7554/eLife.55513.
- Siegel MP, Kruse SE, Percival JM, et al. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell. 2013;12(5):763-771. DOI: 10.1111/acel.12102.
- Campbell MD, Duan J, Samber AT, et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radical Biology and Medicine. 2019;134:268-281. DOI: 10.1016/j.freeradbiomed.2018.12.023.
- Campbell MD, Marthi S, Engles SL, et al. The mitochondrially targeted peptide elamipretide (SS-31) improves ADP sensitivity in aged mitochondria by increasing uptake through the adenine nucleotide translocator (ANT). GeroScience. 2023;45(6):3529-3548. DOI: 10.1007/s11357-023-00861-y.
- Sweetwyne MT, Pippin JW, Eng DG, et al. The mitochondrial-targeted peptide, SS-31, improves glomerular architecture in mice of advanced age. Kidney International. 2017;91(5):1126-1145. DOI: 10.1016/j.kint.2016.10.036.
- Ghonim E, Sabbah HN, Singh-Gupta V, et al. In vivo mitochondrial ATP production is improved in older adult skeletal muscle after a single dose of elamipretide in a randomized trial. PLOS ONE. 2021;16(7):e0253849. DOI: 10.1371/journal.pone.0253849.
- Karaa A, Haas R, Goldstein A, et al. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology. 2018;90(14):e1212-e1221. DOI: 10.1212/WNL.0000000000005255.
- Karaa A, Haas R, Goldstein A, et al. A randomized crossover trial of elamipretide in adults with primary mitochondrial myopathy. Journal of Cachexia, Sarcopenia and Muscle. 2020;11(4):909-918. DOI: 10.1002/jcsm.12559.
- Karaa A, Bertini E, Engelen BGMV, et al. Efficacy and safety of elamipretide in individuals with primary mitochondrial myopathy: the MMPOWER-3 randomized clinical trial. Neurology. 2023;101(3):e238-e252. DOI: 10.1212/WNL.0000000000207402.
- Stebbins KJ, Hughes H, Bhatt RS, et al. Phase 1 clinical trial of elamipretide in intermediate age-related macular degeneration and high-risk drusen: ReCLAIM High-Risk Drusen Study. Ophthalmology Science. 2022;2(2):100095. DOI: 10.1016/j.xops.2021.100095.
- Stebbins KJ, Hughes H, Bhatt RS, et al. Phase 1 clinical trial of elamipretide in dry age-related macular degeneration and noncentral geographic atrophy: ReCLAIM NCGA Study. Ophthalmology Science. 2022;2(2):100096. DOI: 10.1016/j.xops.2021.100096.
- Alam M, Haloi UK, Geng JS, et al. ReCLAIM-2: a randomized phase II clinical trial evaluating elamipretide in age-related macular degeneration. Ophthalmology Science. 2024;4(6):100565. DOI: 10.1016/j.xops.2024.100565.
- Zhang H, Alder NN, Wang W, et al. SS-31, a Mitochondria-Targeting Peptide, Ameliorates Kidney Disease. Oxidative Medicine and Cellular Longevity. 2022;2022:1295509. DOI: 10.1155/2022/1295509.
- Machiraju P, Wang X, Sabouny R, et al. SS-31 peptide reverses the mitochondrial fragmentation present in fibroblasts from patients with DCMA, a mitochondrial cardiomyopathy. Frontiers in Cardiovascular Medicine. 2019;6:167. DOI: 10.3389/fcvm.2019.00167.
- Sabbah HN, Gupta RC, Singh-Gupta V, et al. Elamipretide improves mitochondrial function in the failing human heart. JACC: Basic to Translational Science. 2019;4(3):340-349. DOI: 10.1016/j.jacbts.2019.02.002.
- Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clinical Pharmacology & Therapeutics. 2014;96(6):672-683. DOI: 10.1038/clpt.2014.174.
- Tarantini S, Nyul-Toth A, Giles C, et al. Aging, mitochondrial dysfunction, and cerebral microhemorrhages: a preclinical evaluation of SS-31 (elamipretide). GeroScience. 2025. DOI: 10.1007/s11357-025-01218-x.
- Martinelli D, Catteruccia M, Piemonte F, et al. Elamipretide: a review of its structure, mechanism of action, and therapeutic potential. International Journal of Molecular Sciences. 2025;26(3):944. DOI: 10.3390/ijms26030944.
- Fan M, Luo D, Zhang M, et al. Contemporary insights into elamipretide's mitochondrial mechanism of action and therapeutic effects. Biomedicine & Pharmacotherapy. 2025;184:117859. DOI: 10.1016/j.biopha.2025.117859.
- Li K, Qu Y, Chen Y, et al. Application research of novel peptide mitochondrial-targeted antioxidant SS-31 in mitigating mitochondrial dysfunction. Mitochondrion. 2024;75:101846. DOI: 10.1016/j.mito.2024.101846.
- Stealth BioTherapeutics. FDA Accelerated Approval of FORZINITY (elamipretide) injection for Barth syndrome. Press Release. September 19, 2025.
- FORZINITY (elamipretide) injection. Prescribing Information. Stealth BioTherapeutics Inc. September 2025.