Executive Summary

Key Takeaways
- Bremelanotide (Vyleesi) is the only FDA-approved on-demand treatment for premenopausal HSDD, administered as a 1.75 mg subcutaneous injection
- It works centrally via MC4R activation and dopamine release, not peripherally like PDE5 inhibitors
- RECONNECT trials showed significant improvement in FSFI desire domain scores (effect size 0.49 to 0.61)
- Off-label male studies show promise, especially in PDE5 inhibitor non-responders (34% vs 9% placebo response)
- Most common side effect is nausea (40% first dose), which diminishes with continued use
PT-141, known generically as bremelanotide and marketed under the brand name Vyleesi, is the first and only FDA-approved on-demand treatment for acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women. Unlike conventional erectile dysfunction drugs that work by increasing blood flow to genital tissue, bremelanotide acts through an entirely different pathway - it activates melanocortin-4 receptors (MC4R) in the brain to directly stimulate the neural circuits responsible for sexual desire and arousal.
The U.S. Food and Drug Administration granted approval to bremelanotide in June 2019 following two successful Phase 3 clinical trials known as the RECONNECT studies. These trials enrolled over 1,200 premenopausal women with HSDD and demonstrated that a 1.75 mg subcutaneous injection of bremelanotide, self-administered at least 45 minutes before anticipated sexual activity, produced statistically significant improvements in sexual desire as measured by the Female Sexual Function Index desire domain (FSFI-D) score, along with meaningful reductions in sexually-related personal distress. The drug represented a major step forward because it addressed the central nervous system component of sexual dysfunction rather than simply targeting peripheral vascular responses.
Bremelanotide's origins trace back to research at the University of Arizona in the 1980s, where scientists studying melanocyte-stimulating hormone (MSH) analogs for sunless tanning unexpectedly discovered potent pro-sexual effects. The parent compound, Melanotan II, caused spontaneous erections in nine out of ten male volunteers during early human testing. Palatin Technologies subsequently developed bremelanotide as a metabolite-derived analog of Melanotan II, specifically optimized for sexual dysfunction applications while minimizing tanning and other melanocortin-related side effects.
The drug's mechanism is fundamentally different from PDE5 inhibitors like sildenafil (Viagra) and tadalafil (Cialis). While those medications block the enzyme phosphodiesterase type 5 to enhance blood flow in response to existing arousal, bremelanotide works upstream in the sexual response cascade. It activates MC4 receptors on neurons in the medial preoptic area of the hypothalamus, triggering dopamine release into reward and motivation centers including the nucleus accumbens. This central mechanism means bremelanotide can generate desire itself rather than simply facilitating the physical response to pre-existing desire. For individuals whose sexual dysfunction stems from reduced central drive rather than vascular insufficiency, this distinction is clinically meaningful.
While Vyleesi is approved exclusively for premenopausal women with HSDD, significant off-label interest exists for male sexual dysfunction. Early clinical trials demonstrated that bremelanotide could produce erectile responses in men, including those who had failed PDE5 inhibitor therapy. In a study of 342 sildenafil non-responders, 34% of men receiving bremelanotide achieved erections sufficient for intercourse compared to just 9% on placebo. Palatin Technologies has pursued combination formulations pairing bremelanotide with PDE5 inhibitors for men with refractory erectile dysfunction.
The safety profile of bremelanotide is well-characterized. Nausea is the most common adverse effect, occurring in approximately 40% of patients with the first injection, though this typically diminishes with repeated use. Transient blood pressure elevations averaging 6 mmHg systolic and 3 mmHg diastolic occur and resolve within 12 hours. The drug carries a use limitation of no more than 8 doses per month due to concerns about focal hyperpigmentation with frequent dosing. Bremelanotide is contraindicated in patients with uncontrolled hypertension or cardiovascular disease.
This report provides a thorough examination of PT-141 (bremelanotide), covering its developmental history from Melanotan II, the molecular pharmacology of MC4R activation, detailed RECONNECT trial data, evidence for male off-label use, head-to-head comparison with PDE5 inhibitors, practical dosing and administration guidance, and comprehensive safety information. Whether you are a clinician evaluating treatment options for patients with HSDD or sexual dysfunction, a researcher studying melanocortin pharmacology, or a patient exploring peptide-based therapies through the peptide research hub, this guide synthesizes the current evidence base into actionable clinical intelligence.
Key Takeaways
- Bremelanotide (Vyleesi) is the only FDA-approved on-demand treatment for premenopausal HSDD, administered as a 1.75 mg subcutaneous injection
- It works centrally via MC4R activation and dopamine release, not peripherally like PDE5 inhibitors
- RECONNECT trials showed significant improvement in FSFI desire domain scores (effect size 0.49 to 0.61)
- Off-label male studies show promise, especially in PDE5 inhibitor non-responders (34% vs 9% placebo response)
- Most common side effect is nausea (40% first dose), which diminishes with continued use
- Maximum recommended frequency is 8 doses per month to minimize hyperpigmentation risk
Understanding Hypoactive Sexual Desire Disorder
Before examining bremelanotide in detail, it is worth establishing the clinical context of the condition it treats. HSDD is the most common sexual complaint among women, affecting an estimated 8-10% of premenopausal women in the United States. The condition is defined by persistently or recurrently deficient (or absent) sexual fantasies and desire for sexual activity that causes marked personal distress or interpersonal difficulty. The distress criterion is critical - many women experience periods of low desire without distress, and these women do not meet diagnostic criteria for HSDD.
The pathophysiology of HSDD is complex and likely involves multiple neurobiological factors. Reduced dopaminergic tone in brain reward and motivation centers is one proposed mechanism. Excessive serotonergic inhibition of sexual circuits is another. Hormonal factors, including declining androgen levels, may contribute in some cases. Psychological factors such as stress, body image concerns, relationship dissatisfaction, and past sexual trauma can also play significant roles. In practice, most cases of HSDD likely involve an interplay of biological, psychological, and relational factors.
Prior to bremelanotide's approval, treatment options for HSDD were limited. Flibanserin (Addyi), approved in 2015, was the first pharmacotherapy specifically indicated for HSDD, but it required daily dosing and carried a Risk Evaluation and Mitigation Strategy (REMS) restricting its use with alcohol. Testosterone therapy, while used off-label in some countries, lacks FDA approval for women. Psychological interventions, including cognitive behavioral therapy and mindfulness-based therapies, have evidence of benefit but require sustained engagement and may not address underlying neurobiological deficits.
Bremelanotide filled a distinct clinical niche: an on-demand pharmacotherapy with a novel central mechanism, no alcohol restriction, and no REMS requirement. Its approval expanded the treatment toolkit for clinicians managing HSDD and provided an alternative mechanism of action for patients who do not respond to or cannot tolerate serotonergic therapy.
Clinical Significance and Impact on Sexual Medicine
The approval of bremelanotide has had implications beyond the individual patient level. It validated the melanocortin system as a druggable target for sexual dysfunction, opening the door for future melanocortin-based therapeutics. It also reinforced the concept that female sexual desire is primarily a brain-mediated phenomenon, not a genital blood flow issue, shifting the research paradigm away from the vascular approaches that had repeatedly failed in women.
For the field of sexual medicine, bremelanotide demonstrated that on-demand pharmacotherapy for female desire dysfunction is feasible. The drug showed that the concept of taking a pill (or injection) before anticipated sexual activity, which had long been the model for male erectile dysfunction treatment, could be adapted for female desire disorder. This was a conceptual advancement because HSDD had previously been viewed as a chronic condition requiring continuous (daily) treatment.
The impact on compounding pharmacy practice has also been substantial. Since bremelanotide's FDA approval and the growing clinical evidence base, compounding pharmacies like FormBlends have seen increased demand for PT-141 formulations, particularly for male off-label use and for alternative delivery forms (nasal sprays, sublingual troches) that some patients prefer over injection. This has expanded access to melanocortin-based therapy beyond the narrow FDA-approved indication.
Looking at the broader peptide therapy landscape, bremelanotide joins a growing family of peptide-based treatments that target specific receptor systems with precision. Other peptides available through the peptide research hub include growth hormone secretagogues like CJC-1295/Ipamorelin, tissue repair peptides like BPC-157, and cognitive-enhancing peptides like Semax. Each of these represents a different application of the peptide pharmacology platform, and together they illustrate the versatility of peptide-based therapeutics.
Development from Melanotan II

Figure 2: Development timeline of bremelanotide from early melanocortin research through FDA approval in 2019
The story of bremelanotide begins not with sexual dysfunction research but with the study of skin pigmentation. Understanding this developmental lineage is essential for appreciating both the drug's mechanism and its unique pharmacological profile. The path from tanning peptide to FDA-approved sexual desire treatment spans nearly four decades of research and involves some of the most serendipitous discoveries in modern pharmacology.
The Melanocortin Research Program at the University of Arizona
In the early 1960s, researchers first observed that alpha-melanocyte-stimulating hormone (alpha-MSH) could induce skin darkening in animal models. Alpha-MSH is a 13-amino-acid peptide produced by the pituitary gland that plays a central role in melanogenesis, the biological process by which melanocytes produce the pigment melanin. This natural hormone signals melanocytes to produce more melanin, resulting in darker skin - the same basic process that occurs during sun exposure, but triggered endogenously rather than by ultraviolet radiation.
By the 1980s, scientists at the University of Arizona, led by Dr. Mac Hadley and Dr. Victor Hruby, had embarked on an ambitious program to develop synthetic analogs of alpha-MSH that could serve as sunless tanning agents. The appeal was straightforward: if a drug could stimulate melanin production without UV exposure, it could potentially reduce skin cancer risk while delivering the cosmetic benefit of tanned skin. The researchers synthesized and tested dozens of MSH analogs, searching for compounds with enhanced potency, longer duration of action, and improved stability compared to the natural hormone.
Two standout candidates emerged from this work. The first, [Nle4, D-Phe7]-alpha-MSH, became known as Melanotan I (afamelanotide) and was developed primarily as a tanning agent - it later received approval in the European Union and Australia for patients with erythropoietic protoporphyria, a painful photosensitivity condition. The second candidate, Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2, became known as Melanotan II. This cyclic heptapeptide was a more potent melanocortin receptor agonist with activity across multiple receptor subtypes, and it was this compound that would ultimately lead to bremelanotide.
The Accidental Discovery of Pro-Sexual Effects
During the first human trials of Melanotan II in the early 1990s, researchers observed something entirely unexpected. While the compound did produce the desired skin darkening effect, male volunteers began reporting pronounced sexual side effects. Of the first ten male subjects who received Melanotan II by subcutaneous injection, nine experienced spontaneous erections. This was not a subtle effect - subjects described the erections as occurring without sexual stimulation and persisting for extended periods. Some subjects also reported increased sexual thoughts and heightened arousal.
The serendipity of this discovery cannot be overstated. The research team had been focused exclusively on pigmentation and had no framework for expecting sexual effects. But the melanocortin receptor system, it turned out, plays a dual role - the same MC4 receptors that help regulate pigmentation in the periphery also modulate sexual behavior circuits in the brain. The alpha-MSH analog was crossing the blood-brain barrier and activating hypothalamic neurons involved in sexual motivation and arousal.
One particularly well-documented early case involved a researcher who self-injected a dose of Melanotan II that was later determined to be too high. He experienced severe nausea and vomiting but also a prolonged, painful erection lasting approximately eight hours - a condition known as priapism. This dramatic incident, while medically concerning, provided compelling proof-of-concept that melanocortin agonists could powerfully activate sexual response pathways.
Palatin Technologies Takes the Lead
The sexual dysfunction applications of Melanotan II attracted commercial interest. Competitive Technologies, which held the licensing rights from the University of Arizona, licensed the sexual dysfunction applications to Palatin Technologies, a New Jersey-based biopharmaceutical company. Palatin recognized the commercial potential but also understood that Melanotan II itself had limitations as a drug candidate. The compound's broad melanocortin receptor activity meant it produced tanning, appetite suppression, and other effects alongside the desired pro-sexual activity. It also had stability issues and a complex pharmacokinetic profile.
Palatin ceased development of Melanotan II as a drug candidate in 2000 and instead synthesized a new compound: bremelanotide, designated PT-141. Bremelanotide is a likely metabolite of Melanotan II - specifically, it differs from its parent compound in having a hydroxyl group where Melanotan II has an amide. This structural modification was strategically important. Bremelanotide retained the pro-sexual MC4R agonist activity but had a somewhat different pharmacological profile, and critically, it was a distinct chemical entity that could be independently patented and developed.
Early Clinical Development: The Intranasal Formulation
Palatin initially developed bremelanotide as an intranasal spray, reasoning that this delivery route would offer convenience and rapid onset. Early Phase 1 and Phase 2 studies of the nasal formulation showed promising results in both men and women. In men with erectile dysfunction, intranasal doses of 10 mg and 20 mg produced significant improvements in penile rigidity measurements. In Phase 2 studies in women with female sexual arousal disorder (FSAD), the nasal spray showed statistically significant improvements in arousal and desire scores.
In August 2004, Palatin signed a co-development agreement with King Pharmaceuticals worth $20 million upfront, signaling serious industry confidence in the program. King Pharmaceuticals would share development costs and marketing rights in the United States, while both companies would jointly license the drug internationally.
However, the intranasal program encountered a critical setback. During clinical development, the FDA identified concerns about blood pressure elevations associated with the nasal formulation. The intranasal route of delivery was producing higher peak plasma concentrations than desired, and these spikes correlated with clinically significant increases in blood pressure. In 2007, the FDA placed a clinical hold on all intranasal bremelanotide studies, effectively halting the nasal spray program. King Pharmaceuticals subsequently terminated its partnership with Palatin.
The Pivot to Subcutaneous Injection
Rather than abandoning bremelanotide entirely, Palatin conducted a strategic reassessment. The company's scientists hypothesized that a subcutaneous injection, which produces a more gradual absorption curve than intranasal delivery, might achieve therapeutic plasma concentrations without the problematic blood pressure spikes seen with the nasal route. Preclinical and early clinical data supported this hypothesis.
By 2008, Palatin had pivoted its entire development program to a subcutaneous injection formulation. The lower dose (1.75 mg subcutaneously versus 10-20 mg intranasally) and the more controlled absorption kinetics resulted in a significantly better cardiovascular safety profile. Blood pressure elevations were smaller in magnitude and shorter in duration. The FDA lifted the clinical hold, and Palatin was able to advance into Phase 3 testing.
Palatin also made a strategic decision to focus on female sexual dysfunction rather than male erectile dysfunction. While early studies had shown efficacy in both sexes, the competitive landscape for male ED was already crowded with PDE5 inhibitors (sildenafil, tadalafil, vardenafil, avanafil). Female sexual desire disorder, by contrast, had very few approved treatment options. Flibanserin (Addyi), a daily oral medication working through serotonin receptors, had received FDA approval in 2015 but with a Risk Evaluation and Mitigation Strategy (REMS) due to concerns about hypotension when combined with alcohol. Palatin saw an opportunity to differentiate bremelanotide as an on-demand treatment with a distinct mechanism of action.
Partnership with AMAG Pharmaceuticals and Path to Approval
In 2017, Palatin entered a licensing agreement with AMAG Pharmaceuticals, granting AMAG exclusive North American rights to commercialize bremelanotide. Under the deal, Palatin received $60 million upfront and was eligible for up to $80 million in regulatory and sales milestones plus tiered royalties on net sales. AMAG assumed responsibility for the Phase 3 clinical program and regulatory filings.
AMAG completed two Phase 3 trials - RECONNECT 1 (NCT02333071) and RECONNECT 2 (NCT02338960) - enrolling a combined total of over 1,200 premenopausal women with HSDD. Both studies met their coprimary endpoints, demonstrating statistically significant improvements in sexual desire and reductions in sexual distress. The New Drug Application was filed in March 2018, and on June 21, 2019, the FDA approved Vyleesi (bremelanotide injection) for the treatment of acquired, generalized HSDD in premenopausal women.
The approval marked the culmination of nearly four decades of research, from the initial melanocortin peptide synthesis at the University of Arizona through multiple formulation changes, partnership shifts, and regulatory challenges. It also established a new therapeutic class - melanocortin receptor agonists for sexual dysfunction - that operates through a fundamentally different mechanism than any previously approved sexual health medication. For clinicians and patients interested in the broader peptide research landscape, the peptide research hub provides additional context on how peptide therapeutics have evolved across multiple indications.
Commercialization and Market Position
Following FDA approval, AMAG launched Vyleesi commercially in the fall of 2019. However, the commercial trajectory was challenging. The product's relatively high price point, the requirement for subcutaneous injection (which some patients found less appealing than an oral medication), and the onset of the COVID-19 pandemic in early 2020 all limited uptake. AMAG Pharmaceuticals was acquired by Covis Pharma Group in 2020, and the Vyleesi brand transferred to new ownership.
Meanwhile, the compounding pharmacy market for bremelanotide expanded significantly. Because bremelanotide's peptide structure could be synthesized by qualified compounding pharmacies, clinicians began prescribing compounded versions for both women and men with various forms of sexual dysfunction. Compounded formulations include subcutaneous injections at various doses, nasal sprays, and sublingual troches. This off-label compounding market has become a significant channel for bremelanotide access, particularly for male patients who cannot obtain the FDA-approved Vyleesi product (which is indicated only for premenopausal women).
Palatin Technologies has continued to develop bremelanotide-based therapies. The company has explored combination formulations pairing bremelanotide with PDE5 inhibitors for male erectile dysfunction, and it has investigated oral formulations that could eliminate the need for injection. These ongoing development programs reflect the continued scientific and commercial interest in melanocortin-based approaches to sexual dysfunction treatment.
Development Timeline at a Glance
- 1960s: Alpha-MSH shown to affect pigmentation in animal models
- 1980s: University of Arizona begins synthesizing MSH analogs for tanning
- Early 1990s: Melanotan II produces unexpected pro-sexual effects in human volunteers
- 2000: Palatin Technologies synthesizes bremelanotide (PT-141) as a Melanotan II derivative
- 2004: King Pharmaceuticals partnership ($20M upfront) for intranasal development
- 2007: FDA places clinical hold on intranasal program due to blood pressure concerns
- 2008: Palatin pivots to subcutaneous injection formulation
- 2017: AMAG Pharmaceuticals licenses North American rights ($60M upfront)
- 2019: FDA approves Vyleesi (bremelanotide) for HSDD in premenopausal women
- 2020-present: Ongoing studies of combination therapies and oral formulations
Structural Chemistry of Bremelanotide
Understanding bremelanotide's structure helps explain its pharmacological properties and how it differs from its parent compound Melanotan II. Bremelanotide is a cyclic heptapeptide with the amino acid sequence Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-OH. The "c" notation indicates the cyclic portion of the molecule, formed by a lactam bridge between the Asp and Lys side chains. This cyclization is critical for the compound's stability and receptor binding affinity - the cyclic structure constrains the peptide backbone into a conformation that optimally fits the binding pocket of melanocortin receptors.
The key structural difference from Melanotan II is at the C-terminus: where Melanotan II has an amide group (-NH2), bremelanotide has a free carboxylic acid (-OH). This seemingly minor modification has meaningful pharmacological consequences. The free acid form alters the compound's charge distribution, which affects its receptor binding kinetics and selectivity profile. Bremelanotide retains strong agonist activity at MC3R and MC4R (the receptors most relevant for sexual behavior) while having somewhat modified activity at MC1R (the pigmentation receptor) compared to Melanotan II.
The molecular weight of bremelanotide is approximately 1025 daltons, placing it in the range typical of small peptides. This size is large enough to provide receptor-specific binding but small enough to cross the blood-brain barrier, particularly when administered subcutaneously at therapeutic doses. The drug's ability to access central MC4Rs in the hypothalamus is essential for its pro-sexual effects, and the subcutaneous route provides plasma concentrations sufficient for adequate brain penetration.
The inclusion of D-phenylalanine (D-Phe) at position 7 is another critical structural feature inherited from the Melanotan II backbone. Natural amino acids are almost exclusively in the L-configuration, but the D-amino acid substitution at this position dramatically increases the peptide's resistance to enzymatic degradation. Proteolytic enzymes, which normally break down peptides within minutes in the bloodstream, have difficulty recognizing the non-natural D-amino acid configuration. This gives bremelanotide a plasma half-life of 2.5 hours - long enough for clinical utility but short enough for on-demand dosing without prolonged drug accumulation.
The norleucine (Nle) substitution at position 4 replaces the natural methionine found in alpha-MSH. This change prevents oxidation of the methionine sulfur atom, which would reduce the compound's biological activity and shelf stability. It's a common pharmaceutical chemistry strategy for stabilizing peptide drugs.
The Broader Melanocortin Pharmaceutical Pipeline
Bremelanotide is not the only melanocortin-targeting compound in pharmaceutical development. Several other companies have pursued melanocortin receptor agonists and modulators for various indications, reflecting the therapeutic potential of this receptor system.
Setmelanotide (Imcivree), developed by Rhythm Pharmaceuticals, is an MC4R agonist approved for rare genetic obesity disorders caused by mutations in the POMC, PCSK1, or LEPR genes. Unlike bremelanotide, which is dosed on-demand, setmelanotide is administered daily and targets the appetite-regulating functions of MC4R. Its approval validated MC4R as a therapeutic target for metabolic disease and demonstrated that melanocortin agonists could be used safely with chronic dosing, providing indirect reassurance about long-term melanocortin receptor modulation.
Afamelanotide (Scenesse), the direct descendant of the original Melanotan I peptide, received approval in the EU and Australia for erythropoietic protoporphyria. This compound is a selective MC1R agonist that promotes melanogenesis (tanning) as a photoprotective strategy. Its approval represented a return to the original tanning application of the Arizona melanocortin research program.
Palatin Technologies, the company behind bremelanotide, has continued to develop next-generation melanocortin therapeutics. Their pipeline includes oral melanocortin agonists that could eliminate the need for injection, as well as compounds with improved selectivity profiles designed to minimize off-target effects such as hyperpigmentation and nausea. These development programs reflect the ongoing commercial and scientific interest in melanocortin pharmacology.
For patients and clinicians interested in the broader peptide therapy landscape, these melanocortin program developments sit alongside advances in other peptide families. Growth hormone-releasing peptides like sermorelin and tesamorelin, anti-aging peptides like Epithalon, and metabolic peptides like semaglutide all represent different applications of peptide pharmacology advancing through clinical development and into clinical practice.
MC4R Mechanism of Action
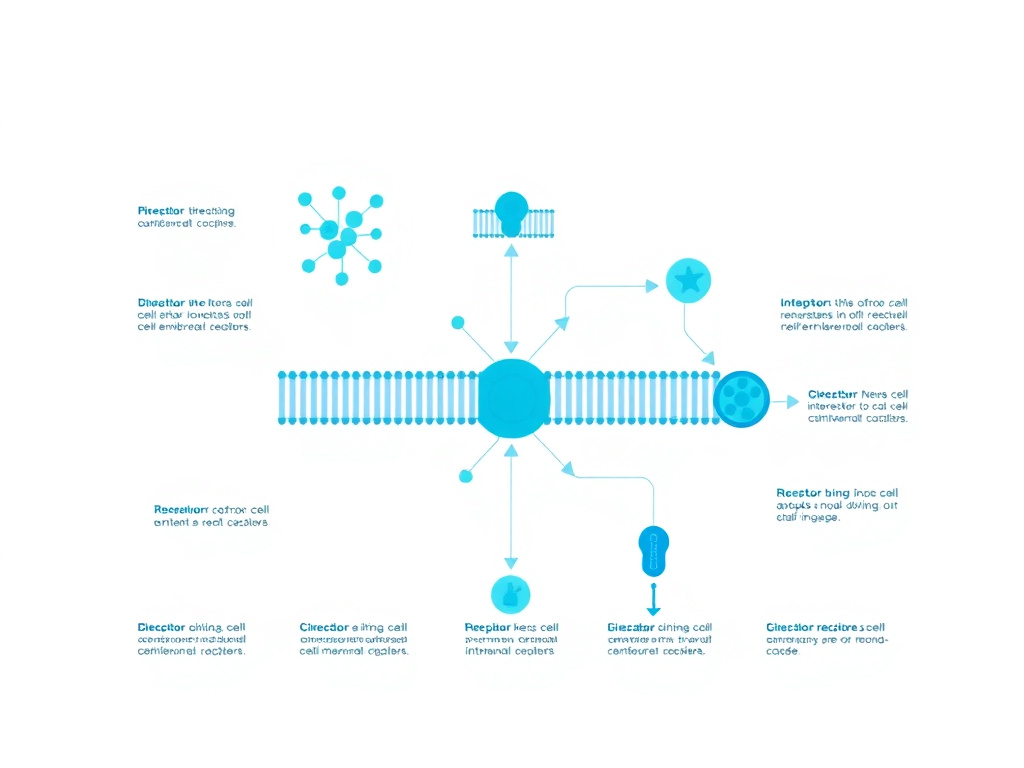
Bremelanotide's mechanism of action is fundamentally distinct from every other approved treatment for sexual dysfunction. While PDE5 inhibitors target peripheral blood flow, and flibanserin modulates serotonin receptor activity, bremelanotide directly activates the melanocortin receptor system - one of the brain's primary pathways for regulating motivated behavior, including sexual desire and arousal. Understanding this mechanism requires a journey through melanocortin receptor pharmacology, hypothalamic neuroanatomy, and the neuroscience of sexual motivation.
The Melanocortin Receptor Family
The melanocortin receptor system consists of five G-protein coupled receptor subtypes, designated MC1R through MC5R. Each subtype has distinct tissue distribution patterns and physiological roles. MC1R is predominantly expressed in melanocytes and mediates skin pigmentation responses. MC2R is the ACTH receptor found in the adrenal cortex. MC3R and MC4R are expressed primarily in the central nervous system and play roles in energy homeostasis, metabolism, and behavior. MC5R is found in exocrine glands and peripheral tissues.
Bremelanotide is a non-selective melanocortin receptor agonist with activity at MC1R, MC3R, MC4R, and MC5R (but not MC2R). Its pro-sexual effects are attributed primarily to MC4R activation, though MC3R may play a contributing role. The drug's activity at MC1R explains why prolonged or frequent dosing can produce skin hyperpigmentation - a remnant of its origins as a derivative of a tanning peptide. This receptor profile also explains the drug's effects on nausea (melanocortin receptors in the brainstem's area postrema) and transient blood pressure changes (central sympathetic activation).
The endogenous ligands for melanocortin receptors include alpha-MSH, beta-MSH, gamma-MSH, and ACTH, all derived from the precursor protein proopiomelanocortin (POMC). In the brain, alpha-MSH is the primary melanocortin signal acting at MC4R. POMC-expressing neurons in the arcuate nucleus of the hypothalamus project widely throughout the brain, releasing alpha-MSH to regulate feeding behavior, energy expenditure, cardiovascular function, and - critically for bremelanotide's therapeutic application - sexual behavior.
MC4R and the Neural Circuitry of Sexual Desire
MC4R is abundantly expressed in several brain regions that constitute the core neural circuit for sexual motivation and behavior. The most critical of these is the medial preoptic area (mPOA) of the hypothalamus. The mPOA has been recognized for decades as a key integration center for sexual behavior in both males and females across mammalian species. Lesion studies in rodents show that damage to the mPOA eliminates or severely impairs copulatory behavior, while electrical stimulation of this region facilitates sexual responses.
Beyond the mPOA, MC4R is expressed in the ventral tegmental area (VTA), the nucleus accumbens, the amygdala, and the paraventricular nucleus of the hypothalamus (PVN). These regions collectively form a motivational circuit that links sexual stimuli to reward, arousal, and behavioral output. The VTA is the origin of the mesolimbic dopamine pathway - the brain's primary reward and motivation circuit. The nucleus accumbens receives dopaminergic input from the VTA and translates motivational signals into behavioral drive. The amygdala processes emotional and social cues relevant to sexual contexts. The PVN coordinates autonomic and endocrine responses to sexual stimulation, including oxytocin release.
Animal studies have elucidated the specific mechanism by which MC4R activation promotes sexual behavior. When bremelanotide binds to presynaptic MC4Rs on neurons in the mPOA, it triggers increased release of dopamine. This dopamine then acts on downstream targets in the nucleus accumbens and other reward regions, generating the subjective experience of sexual desire and the motivational drive to seek sexual activity. In female rodent models, MC4R agonists increase solicitation behaviors (proceptivity) and receptive postures (lordosis), while MC4R antagonists block these behaviors even in hormonally primed animals.
The Dopamine Connection
Dopamine is the brain's primary neurotransmitter for motivation, reward anticipation, and goal-directed behavior. Its role in sexual desire is well-established across multiple lines of evidence. Dopamine-enhancing drugs like apomorphine and L-DOPA are known to increase sexual drive, while dopamine-blocking antipsychotics commonly cause sexual dysfunction as a side effect. The mesolimbic dopamine system - projecting from the VTA to the nucleus accumbens - is activated by sexual stimuli and during sexual activity in both human neuroimaging studies and animal electrophysiology recordings.
Bremelanotide's pro-sexual effect can be understood as a pharmacological amplification of this natural dopamine-mediated desire circuit. By activating MC4Rs that facilitate dopamine release, bremelanotide essentially turns up the volume on the brain's sexual motivation signal. This is why the drug can generate desire in the absence of external sexual stimulation - it is acting upstream of the normal stimulus-response pathway, directly enhancing the neurochemical signal that the brain interprets as wanting.
This dopaminergic mechanism also explains bremelanotide's efficacy in conditions where desire itself is impaired. In HSDD, the fundamental problem is not the inability to respond to arousal (as in erectile dysfunction) but rather the absence or insufficiency of the desire signal that would normally initiate the sexual response cascade. By boosting dopamine release in motivation and reward circuits, bremelanotide addresses the core neurochemical deficit underlying HSDD. For individuals whose low desire stems from central dopaminergic insufficiency rather than hormonal, relational, or psychological factors, this mechanism is particularly well-suited.
fMRI Evidence: Bremelanotide Changes Brain Activation Patterns
Some of the most compelling evidence for bremelanotide's central mechanism comes from functional magnetic resonance imaging (fMRI) studies conducted in women with HSDD. A study published in the Journal of Clinical Investigation examined brain activation patterns in women with HSDD before and after bremelanotide administration, compared to healthy controls and placebo.
The researchers found that women with HSDD showed reduced activation in reward and motivation brain regions when viewing erotic stimuli compared to healthy controls. This hypoactivation pattern was consistent with the clinical symptom of reduced desire - the brain's motivational circuits were simply not responding adequately to sexual cues. Following bremelanotide administration, activation in these regions increased significantly, approaching the levels seen in healthy women.
Perhaps more telling, bremelanotide also modulated functional connectivity patterns. Women with HSDD showed reduced connectivity between prefrontal cortical regions and subcortical reward areas. This disconnection may represent a form of top-down suppression of sexual motivation. Bremelanotide appeared to restore this connectivity, suggesting that the drug may work not only by boosting bottom-up motivational signals but also by reducing top-down inhibitory processes that suppress desire.
These neuroimaging findings provide a biological correlate for the subjective experience reported by women in clinical trials: not just increased sexual desire, but a reduction in the mental effort required to become interested in and engaged with sexual activity. The drug appears to shift the brain's baseline state from one of relative sexual disinterest to one of readiness and receptivity.
The Broader Melanocortin Signaling Context
MC4R signaling does not operate in isolation. It interacts with multiple other neurotransmitter and neuropeptide systems that influence sexual behavior. Oxytocin, which is released from the PVN during sexual activity and promotes pair bonding and social connection, is regulated in part by melanocortin signaling. Oxytocin and melanocortin systems appear to have bidirectional interactions, with MC4R activation facilitating oxytocin release and oxytocin in turn modulating melanocortin neuron activity.
The opioid system also intersects with melanocortin sexual behavior pathways. Beta-endorphin, which is co-released with alpha-MSH from POMC neurons, acts on mu-opioid receptors to produce pleasure and reward. The balance between melanocortin (pro-sexual, activating) and opioid (post-consummatory, inhibiting) signals from POMC neurons may help regulate the sexual response cycle from desire through arousal, orgasm, and resolution.
Additionally, gonadal hormones modulate MC4R expression and signaling. Estrogen upregulates MC4R in the mPOA and enhances behavioral responses to melanocortin agonists in female rodent models. This hormonal interaction may partly explain why bremelanotide's clinical efficacy has been most clearly demonstrated in premenopausal women with normal hormonal profiles. It also suggests that hormonal status may influence individual response to the drug, a consideration relevant for patients using hormonal contraceptives or undergoing hormone therapy. For those interested in how hormonal pathways intersect with peptide therapy, the kisspeptin-10 product page provides additional context on hypothalamic-pituitary-gonadal axis signaling.
Why MC4R Agonism Differs from Other Approaches
The MC4R mechanism of bremelanotide distinguishes it from all other pharmacological approaches to sexual dysfunction in several fundamental ways. First, it acts upstream in the sexual response cascade, targeting desire rather than physical response. Second, it engages a specific motivational circuit rather than broadly affecting neurotransmitter tone (as does flibanserin's serotonergic mechanism). Third, its on-demand pharmacokinetics mean that MC4R activation occurs transiently and can be timed to coincide with desired sexual activity, rather than requiring chronic daily dosing.
These distinctions have practical clinical implications. A patient who has adequate vascular function but lacks sexual desire will likely respond better to bremelanotide than to a PDE5 inhibitor. Conversely, a patient with strong desire but inability to achieve or maintain an erection due to vascular disease will benefit more from PDE5 inhibition. Understanding where in the sexual response cascade the dysfunction occurs is key to selecting the appropriate pharmacological intervention.
For clinicians and patients exploring the full spectrum of peptide-based interventions for sexual health, the PT-141 product page provides formulation and ordering information, while the dosing calculator can help individualize therapy.
Clinical Relevance
The MC4R mechanism means bremelanotide works best for patients whose primary complaint is low desire rather than physical inability to perform. It addresses the motivational and psychological component of sexual dysfunction at its neurochemical root. Patients who report "I can function physically but I just don't feel like it" are the ideal candidates for MC4R agonist therapy. This is in contrast to PDE5 inhibitors, which are best suited for patients who say "I want to but I physically can't."
Implications for Personalized Sexual Medicine
The MC4R mechanism of bremelanotide has important implications for personalized approaches to sexual dysfunction treatment. Not all sexual desire problems have the same underlying neurobiology, and understanding the specific pathway disrupted in an individual patient can guide treatment selection. A patient with reduced dopaminergic tone in motivation circuits (potentially identifiable through functional neuroimaging or clinical phenotyping) may be an ideal candidate for MC4R agonist therapy. In contrast, a patient whose low desire stems from excessive serotonergic inhibition might respond better to a serotonin-modulating approach like flibanserin.
Genetic factors may also influence response to bremelanotide. Polymorphisms in the MC4R gene have been identified that affect receptor function and are associated with variations in body weight, energy metabolism, and potentially sexual behavior. While genetic testing for MC4R variants is not yet part of routine clinical practice for sexual dysfunction, the concept of pharmacogenomic matching - selecting medications based on an individual's genetic receptor profile - represents a future direction for the field.
Hormonal status is another personalization factor. As noted, estrogen modulates MC4R expression in the hypothalamus. Women with adequate estrogen levels may have higher MC4R density and stronger responses to bremelanotide compared to those with lower estrogen (such as postmenopausal women or those using hormonal contraceptives that suppress ovarian estrogen production). This interaction between hormonal milieu and melanocortin receptor function is an active area of investigation.
The age-related decline in dopaminergic function is yet another factor that could influence bremelanotide response. Dopamine receptor density and dopamine synthesis capacity both decrease with aging, which may contribute to age-related declines in sexual desire. Bremelanotide's ability to facilitate dopamine release could theoretically compensate for some of this age-related dopaminergic decline, but the degree of compensation may depend on the residual capacity of the dopamine system.
For clinicians navigating these complexities, the dosing calculator provides a starting framework for individualized therapy, and the free assessment can connect patients with providers experienced in personalized peptide-based treatment planning.
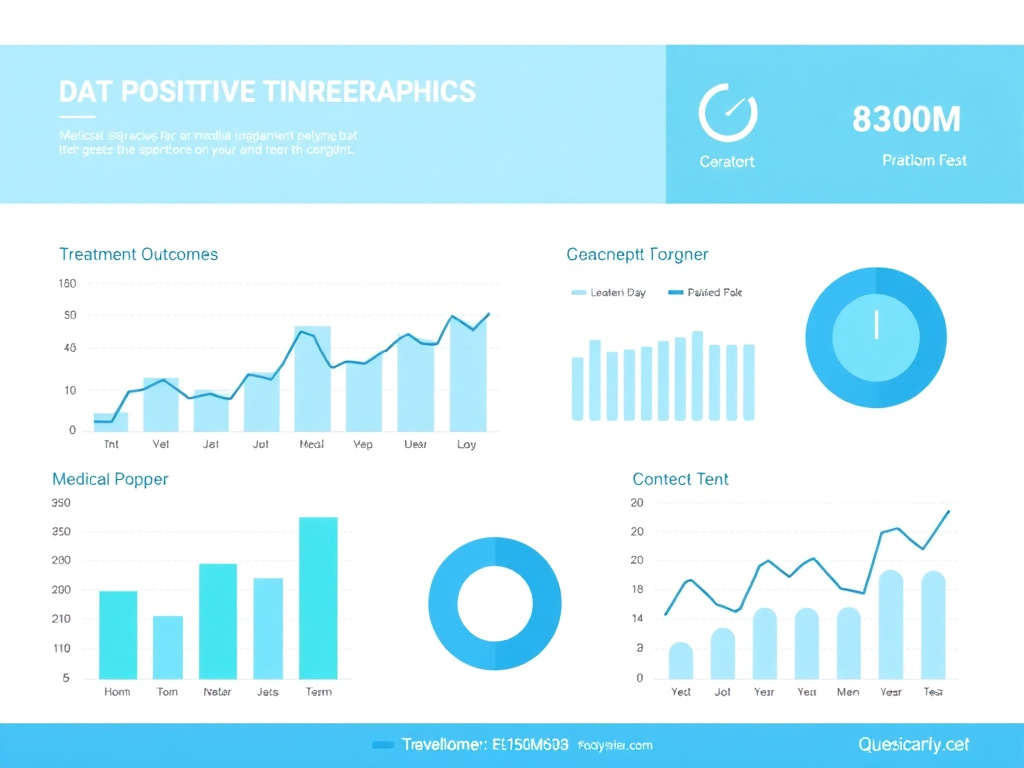
RECONNECT Trial Results
The RECONNECT trials represent the definitive Phase 3 evidence supporting bremelanotide's FDA approval. These two identical, randomized, double-blind, placebo-controlled studies were the largest and most rigorously designed clinical evaluations of any melanocortin agonist for sexual dysfunction. Their results established bremelanotide as an effective, on-demand treatment for HSDD and provided the data package that convinced the FDA to approve Vyleesi.
Study Design and Patient Population
The RECONNECT program consisted of two parallel Phase 3 studies: Study 301 (NCT02333071) and Study 302 (NCT02338960). Both studies used identical protocols, a deliberate design choice that allowed for both individual study analysis and integrated pooled analysis, strengthening the overall evidence base.
Each study enrolled premenopausal women aged 21 and older with acquired, generalized HSDD of at least 6 months' duration. The diagnosis was established according to the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision (DSM-IV-TR) criteria. Key inclusion criteria required that patients have a stable relationship of at least 6 months and that their HSDD was not attributable to co-existing medical or psychiatric conditions, relationship issues, medication effects, or substance use.
Patients were randomized 1:1 to receive either bremelanotide 1.75 mg or placebo, administered by subcutaneous injection as needed before anticipated sexual activity. The treatment period was 24 weeks, with patients using up to 8 doses per month. Both studies included a 4-week run-in period to establish baseline measures.
In total, 1,247 women were randomized across the two studies: 634 to bremelanotide and 613 to placebo. The mean age was approximately 39 years. The population was predominantly Caucasian (approximately 85%), with representation from Black, Hispanic, and other racial and ethnic groups. Patients had a mean HSDD duration of approximately 4.5 years at baseline, indicating a chronic condition with substantial clinical burden.
Coprimary Endpoints
The RECONNECT trials used two coprimary efficacy endpoints, both of which needed to reach statistical significance for the studies to be considered positive:
1. Change in FSFI Desire Domain Score: The Female Sexual Function Index (FSFI) is a validated 19-item questionnaire assessing six domains of female sexual function: desire, arousal, lubrication, orgasm, satisfaction, and pain. The desire domain specifically measures the frequency and level of sexual desire over the preceding 4 weeks. Scores range from 1.2 to 6.0, with higher scores indicating better function. The FSFI-D was chosen as a coprimary endpoint because it directly measures the core symptom of HSDD - reduced sexual desire.
2. Change in FSDS-DAO Item 13: The Female Sexual Distress Scale-Desire/Arousal/Orgasm (FSDS-DAO) measures sexually related personal distress. Item 13 specifically asks about distress related to low sexual desire. This distress component was included as a coprimary endpoint because HSDD is defined not merely by low desire but by low desire that causes marked personal distress. A treatment that increased desire without reducing distress, or that reduced distress without increasing desire, would be considered incomplete.
Efficacy Results: Individual Studies
Study 301 Results: In the first RECONNECT trial, bremelanotide produced a statistically significant improvement in FSFI desire domain score compared to placebo. The adjusted mean change from baseline was +0.5 points greater with bremelanotide than with placebo (treatment difference, 95% CI: statistically significant, p less than 0.001). On the FSDS-DAO Item 13, bremelanotide also showed significant improvement versus placebo.
Study 302 Results: The second RECONNECT trial replicated the findings of Study 301. The FSFI desire domain improvement was numerically larger than in Study 301, with a treatment difference versus placebo of approximately +0.6 points (p less than 0.001). The FSDS-DAO Item 13 results were similarly positive.
Integrated Analysis
When the data from both RECONNECT studies were pooled, bremelanotide demonstrated a mean improvement in FSFI desire domain score of +0.35 points greater than placebo (p less than 0.001). The effect size ranged from 0.49 to 0.61 across the two studies, indicating a moderate and clinically meaningful treatment effect. To put this in perspective, an effect size of 0.5 is conventionally considered a medium effect in behavioral research and exceeds the threshold typically used to establish clinical relevance in sexual dysfunction studies.
RECONNECT Trial: Change in FSFI Desire Domain Score
The integrated analysis of FSDS-DAO Item 13 showed that bremelanotide reduced sexually related distress by a significantly greater magnitude than placebo. Women on bremelanotide reported feeling less bothered by their low desire, less frustrated, and less distressed about its impact on their relationships and self-esteem.
Secondary Endpoints and Additional Analyses
Beyond the coprimary endpoints, the RECONNECT trials evaluated several secondary measures that provide additional context about bremelanotide's effects:
FSFI Total Score: Bremelanotide improved the overall FSFI total score, reflecting improvement not only in desire but also in arousal, orgasm, and satisfaction domains. This was consistent with the understanding that desire and arousal are closely linked and that improvements in desire tend to cascade through the subsequent stages of the sexual response cycle.
Satisfying Sexual Events (SSEs): The number of satisfying sexual events per month increased modestly with bremelanotide compared to placebo. While the raw numerical increase was small (approximately 0.5 additional SSEs per month), this represented a proportionally meaningful change given the low baseline rates in this population.
Elements of Desire Questionnaire (EDQ): This validated measure assesses multiple components of sexual desire, including spontaneous desire, responsive desire, and sexual motivation. Bremelanotide showed improvements across EDQ domains, suggesting that the drug enhanced multiple facets of desire rather than affecting only a single dimension.
Patient Global Impression of Change (PGIC): A significantly greater proportion of bremelanotide-treated women rated their condition as "much improved" or "very much improved" compared to placebo-treated women, supporting the clinical relevance of the statistically measured changes.
Responder Analysis
Responder analyses revealed that approximately 35% of women receiving bremelanotide achieved a clinically meaningful response (defined as a 0.6-point or greater improvement in FSFI desire domain score) compared to approximately 22% on placebo - an absolute difference of 13 percentage points and a relative improvement of approximately 60%. While these numbers indicate that bremelanotide does not work for every woman with HSDD, the responder rate significantly exceeded placebo and was consistent across both studies.
Subgroup analyses explored whether efficacy varied by age, race, baseline severity, hormonal contraceptive use, or other factors. Generally, bremelanotide's efficacy was consistent across subgroups, though there was a trend toward larger treatment effects in women with more severe baseline HSDD, suggesting that the drug may be particularly useful in moderate-to-severe cases where the unmet need is greatest.
Time Course of Response
Improvements in FSFI desire domain scores were apparent as early as 4 weeks into treatment and continued to accrue through the 24-week study period. The distress reduction (FSDS-DAO) showed a similar time course, with early improvements that were maintained through the end of the study. This pattern suggests that while some women experience benefit relatively quickly, continued use over several months may yield additional improvement.
The mean number of bremelanotide injections used per month was approximately 2.5 to 3, indicating that most women did not use the drug at the maximum permitted frequency of 8 doses per month. This usage pattern suggests that women self-titrated their use based on anticipated sexual activity and personal response, consistent with the drug's intended role as an on-demand treatment.
Open-Label Extension Study
Following the 24-week double-blind phase, patients who completed the RECONNECT studies had the option to enroll in a 52-week open-label extension (OLE) study. This extension provided important data on the long-term efficacy and safety of bremelanotide in real-world use patterns.
In the OLE study, improvements in FSFI desire scores and FSDS-DAO distress scores observed during the double-blind phase were maintained throughout the extension period. There was no evidence of tolerance (loss of efficacy with continued use) over the course of up to 76 weeks of total exposure. Women who had been on placebo during the double-blind phase and switched to bremelanotide in the OLE showed improvements comparable to those seen in the original treatment group, confirming that the drug's effects were not limited to initial exposure.
The OLE data were published by Simon et al. in Obstetrics & Gynecology (2019) and provided reassurance that bremelanotide could be used safely and effectively over extended periods. This was a key consideration for the FDA, given that HSDD is typically a chronic condition requiring ongoing treatment.
Limitations and Interpretation
While the RECONNECT results were positive and supported FDA approval, several limitations deserve acknowledgment. The study population was predominantly Caucasian, and data on efficacy in other racial and ethnic groups were limited. The trials excluded women with significant medical comorbidities, psychiatric conditions, or relationship problems, meaning the results may not generalize to more complex clinical presentations.
The placebo response in sexual dysfunction trials is typically substantial, and the RECONNECT trials were no exception - placebo-treated women also showed improvements, reflecting the well-known effects of study participation, attention, and expectation on sexual function outcomes. However, bremelanotide consistently exceeded placebo by a statistically and clinically significant margin across both studies and multiple endpoints.
The trials also did not include direct comparisons with other HSDD treatments such as flibanserin. Head-to-head comparison data would be valuable for guiding clinical decision-making but are not currently available from controlled trials.
For clinicians and patients weighing bremelanotide therapy, the RECONNECT data support its use as an effective, on-demand treatment for HSDD in premenopausal women, with a moderate effect size that exceeds placebo and is maintained over long-term use. The GLP-1 research hub covers related metabolic and weight management topics for patients with overlapping concerns.
Interpreting the Numbers
An effect size of 0.49 to 0.61 on the FSFI desire domain means that, on average, a woman taking bremelanotide will experience a noticeable increase in sexual desire compared to one taking placebo. The 35% responder rate (vs 22% on placebo) translates to approximately 1 in 3 women experiencing meaningful improvement. The number needed to treat (NNT) is approximately 8, meaning that for every 8 women treated with bremelanotide, 1 will achieve a clinically meaningful response who would not have done so with placebo alone.
Comparison with Flibanserin Trial Data
While no head-to-head trials have directly compared bremelanotide and flibanserin, comparing their Phase 3 datasets provides useful context for clinical decision-making. Flibanserin was evaluated in three Phase 3 trials (VIOLET, DAISY, and BEGONIA) enrolling a combined total of over 2,400 premenopausal women with HSDD. The coprimary endpoints were similar to the RECONNECT trials: FSFI desire domain score and FSDS-R (Female Sexual Distress Scale-Revised) Item 13.
In the flibanserin trials, the mean improvement in satisfying sexual events (SSEs) was approximately 0.5 to 1.0 per month more than placebo. The FSFI desire domain improvement was statistically significant but modest (approximately 0.3 to 0.4 points greater than placebo). The overall effect size was comparable to or slightly smaller than what was observed with bremelanotide in the RECONNECT trials.
Key differences between the two drugs' clinical profiles include:
| Feature | Bremelanotide (Vyleesi) | Flibanserin (Addyi) |
|---|---|---|
| Dosing | On-demand (1.75 mg SC injection) | Daily (100 mg oral) |
| Mechanism | MC4R agonist (dopaminergic) | 5-HT1A agonist / 5-HT2A antagonist |
| Onset | 45-60 minutes | 4-8 weeks for full effect |
| REMS | No | Yes (alcohol interaction) |
| Alcohol restriction | None | Must avoid alcohol (hypotension risk) |
| Route | Subcutaneous injection | Oral tablet (at bedtime) |
| Main side effect | Nausea (40%) | Dizziness (11%), somnolence (11%) |
| Effect size (FSFI-D) | 0.49-0.61 | 0.3-0.5 (estimated) |
The on-demand versus daily dosing distinction is perhaps the most clinically relevant difference for patients. Women who prefer not to take a daily medication for a condition that affects them intermittently may favor bremelanotide's as-needed model. Conversely, women who dislike injections or who want continuous support for desire may prefer flibanserin's oral daily approach. The alcohol restriction on flibanserin is also a significant practical consideration - women who consume alcohol regularly may find this restriction burdensome, making bremelanotide a more convenient option.
Real-World Effectiveness versus Clinical Trial Efficacy
Clinical trial results represent efficacy under controlled conditions - carefully selected patients, monitored compliance, and structured follow-up. Real-world effectiveness often differs from clinical trial efficacy for several reasons. Patients in clinical practice are more diverse than trial populations, compliance may be lower without the structure of a study protocol, and expectations may differ.
Early real-world experience with Vyleesi has provided some data on how the drug performs outside the trial setting. Reports from sexual medicine clinics suggest that responder rates in clinical practice are generally consistent with the RECONNECT trial results, though individual experiences vary widely. Some women report dramatic improvements in desire and sexual satisfaction, while others notice modest or no benefit.
Several factors appear to predict real-world response:
- Baseline severity: Women with more severe HSDD (very low baseline desire) may show larger absolute improvements
- Expectations: Realistic expectations about the drug's effects (moderate improvement in desire, not a transformation) are associated with greater satisfaction
- Nausea tolerance: Women who can tolerate the first few episodes of nausea long enough to develop tachyphylaxis tend to continue therapy
- Relationship context: Women in supportive relationships where both partners understand the condition and the treatment tend to have better outcomes
- Timing: Women who learn to optimize injection timing relative to anticipated sexual activity report better results
Clinicians prescribing bremelanotide should counsel patients that the drug is not an instant transformation but rather a tool that can shift the balance toward greater desire when used in the context of otherwise healthy sexual functioning. Setting appropriate expectations at the outset improves adherence and satisfaction. The free assessment at FormBlends can help patients determine whether bremelanotide is appropriate for their specific situation.
Male Sexual Dysfunction Research

While bremelanotide's FDA approval is limited to HSDD in premenopausal women, some of the earliest and most intriguing clinical evidence for the compound came from studies in men with erectile dysfunction. These male studies, though they never progressed to Phase 3 registration trials, provide valuable data on bremelanotide's potential for male sexual dysfunction and have fueled a significant off-label prescribing practice through compounding pharmacies.
Phase 1 Rigidity Studies in Healthy Volunteers
The earliest human data on bremelanotide's erectile effects came from Phase 1 studies using RigiScan monitoring, a device that measures penile rigidity and tumescence during sleep or in response to visual sexual stimulation. In a double-blind, placebo-controlled study of 32 healthy male volunteers, intranasal bremelanotide at doses of 10 mg and 20 mg produced significant increases in the duration of penile base rigidity at or above 80% - the threshold considered sufficient for vaginal penetration.
At the 20 mg intranasal dose, the mean duration of base rigidity exceeding 80% was approximately 24 minutes, with onset occurring about 30 minutes after administration. This was significantly greater than the placebo response and established proof-of-concept that melanocortin activation could produce measurable erectile responses in human males. The finding was consistent with the earlier Melanotan II observations in which male volunteers experienced spontaneous erections during tanning studies.
What made these results particularly interesting was that the erectile responses occurred in healthy men without erectile dysfunction, and in some cases without concurrent sexual stimulation. This suggested that bremelanotide could generate erectile responses through central mechanisms independent of the peripheral arousal pathway, a finding with important implications for patients whose erectile dysfunction has a psychological or neurological rather than vascular origin.
Phase 2A Studies in Erectile Dysfunction
Following the Phase 1 results, Palatin conducted Phase 2A studies specifically in men diagnosed with erectile dysfunction. These studies used the intranasal formulation at doses ranging from 7 mg to 20 mg. In men with mild-to-moderate ED, bremelanotide significantly improved erectile function scores on the International Index of Erectile Function (IIEF) questionnaire compared to placebo.
The studies also used RigiScan monitoring to objectively document erectile responses. Men with ED who received bremelanotide showed increased rigidity episodes and longer duration of rigidity compared to placebo. The onset of effect was approximately 30 to 60 minutes, comparable to the time course of PDE5 inhibitors.
But what truly distinguished bremelanotide from existing ED treatments was its efficacy in men who had not responded adequately to PDE5 inhibitors. This was clinically significant because an estimated 30-40% of men with ED do not achieve satisfactory results with sildenafil, tadalafil, or other PDE5 inhibitors. For these men, treatment options are limited to injectable prostaglandins (alprostadil), vacuum erection devices, or surgical penile implants - all of which are more invasive, less convenient, and less satisfying than oral or injectable medications.
The PDE5 Inhibitor Non-Responder Study
The most clinically meaningful study of bremelanotide in men was a randomized, double-blind, placebo-controlled trial specifically enrolling men who had failed sildenafil therapy. This study, published in the Journal of Urology, enrolled 342 men with documented erectile dysfunction who had tried and failed sildenafil at adequate doses.
Subjects received either intranasal bremelanotide (10 mg) or placebo before sexual encounters. The primary endpoint was whether subjects could achieve an erection sufficient for vaginal intercourse. The results were striking: 51 subjects (33.5%) in the bremelanotide group achieved successful intercourse compared to only 13 subjects (8.5%) in the placebo group. This represented a nearly fourfold improvement in the success rate among men who had already failed first-line PDE5 inhibitor therapy.
The difference was statistically significant (p less than 0.001) and clinically meaningful. For a patient population with limited remaining options, a 34% success rate represented a tangible improvement in quality of life. The study demonstrated that bremelanotide's central mechanism of action could activate the sexual response in men for whom peripheral vascular therapy was insufficient - evidence that the disconnect between brain and body in sexual dysfunction can sometimes be bridged by addressing the central component.
Combination Therapy: Bremelanotide Plus PDE5 Inhibitors
Recognizing that bremelanotide's central mechanism and PDE5 inhibitors' peripheral mechanism target different components of the erectile pathway, researchers explored the hypothesis that combining both approaches might produce additive benefits. The rationale was straightforward: bremelanotide could enhance central sexual motivation and arousal signals, while a PDE5 inhibitor could ensure that these signals were effectively translated into the peripheral vascular response needed for erection.
A study published examining the combination of bremelanotide with sildenafil in men who were partial responders to sildenafil alone found that the combination produced clinically significant enhanced erectile responses compared to either agent alone. Men who had only marginal benefit from sildenafil experienced substantially improved erectile quality and satisfaction when bremelanotide was added.
Palatin Technologies has continued to pursue this combination strategy. The company developed a co-formulation combining bremelanotide with a PDE5 inhibitor in a single injection, designed for convenience and optimized pharmacokinetics. Phase 2 studies of this combination product in PDE5 inhibitor non-responders have been conducted, with the company reporting promising preliminary data.
Bremelanotide for Male Hypoactive Sexual Desire
Beyond erectile dysfunction, there is growing recognition that male hypoactive sexual desire disorder (MHSDD) is a distinct clinical entity that is underdiagnosed and undertreated. While MHSDD does not yet have the same level of diagnostic codification as female HSDD, clinicians increasingly encounter men whose primary complaint is reduced sexual interest and motivation rather than erectile inability.
Bremelanotide's central mechanism of action makes it theoretically well-suited for MHSDD, since the drug directly enhances dopaminergic motivation circuits. Anecdotal clinical reports from physicians prescribing compounded bremelanotide for men have described improvements not only in erectile function but also in sexual desire, anticipation, and overall sexual satisfaction. However, controlled clinical trial data specifically evaluating bremelanotide for male desire disorder are limited.
The off-label use of PT-141 for male sexual dysfunction has become a significant practice in integrative and sexual medicine clinics. Compounding pharmacies prepare bremelanotide in various formulations for male patients, including subcutaneous injections (typically at 1 to 2 mg doses), sublingual troches, and nasal sprays. Clinicians who prescribe compounded bremelanotide for men typically target patients who have failed or are intolerant of PDE5 inhibitors, who have low desire as a primary complaint, or who have mixed erectile and desire dysfunction.
Special Male Populations
Post-Prostatectomy Patients: Men who have undergone radical prostatectomy for prostate cancer frequently experience erectile dysfunction due to damage to the cavernous nerves that run along the prostate. These patients often respond poorly to PDE5 inhibitors because the neural pathway needed to initiate the nitric oxide-mediated erection response has been disrupted. Bremelanotide's central mechanism, which does not depend on intact peripheral innervation, offers theoretical promise for this population, though controlled trial data are lacking.
Psychogenic Erectile Dysfunction: Men whose erectile dysfunction is primarily psychological rather than vascular are another population of interest for bremelanotide. Performance anxiety, depression, and relationship stress can all inhibit the central neural signals that initiate erection. By boosting dopaminergic motivation and reducing the threshold for sexual arousal, bremelanotide may help overcome the psychological barriers that prevent adequate sexual response. Early clinical experience and small case series have reported encouraging results in this population.
Antidepressant-Induced Sexual Dysfunction: Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) are among the most commonly prescribed medications worldwide, and sexual dysfunction is one of their most common and distressing side effects. SSRI-induced sexual dysfunction typically involves reduced desire, delayed orgasm, and in some cases erectile difficulty. Because SSRIs primarily affect serotonergic neurotransmission and can indirectly inhibit dopaminergic activity, bremelanotide's dopamine-facilitating mechanism may help counteract SSRI-related sexual suppression. Preliminary clinical observations support this hypothesis, though no controlled trials have been completed.
Testosterone Deficiency: Men with low testosterone who do not fully respond to testosterone replacement therapy may benefit from bremelanotide as an adjunctive treatment. Testosterone supports baseline sexual desire and function, but some men on adequate testosterone replacement still report suboptimal desire. Adding a melanocortin agonist to address the downstream dopaminergic component of desire could provide additional benefit. This approach remains empirical, and formal clinical validation is needed.
Regulatory Pathway and Future Prospects for Male Indication
As of 2026, no company has filed for FDA approval of bremelanotide for any male indication. The regulatory pathway would require Phase 3 trials demonstrating efficacy and safety in a defined male population, which represents a substantial investment. Palatin Technologies has expressed interest in pursuing male indications, particularly with its combination formulation, but a registration-quality Phase 3 program has not yet been initiated.
In the meantime, male patients access bremelanotide through compounding pharmacies under physician prescription. This off-label use is legal and increasingly common, but patients should be aware that compounded medications have not undergone the same regulatory review as FDA-approved products. Working with a knowledgeable physician and a reputable compounding pharmacy, such as those supplying FormBlends PT-141, is important for ensuring quality and appropriate dosing.
Off-Label Use Considerations
Bremelanotide is not FDA-approved for any indication in men. All male use is off-label. While off-label prescribing is legal and represents standard medical practice when supported by clinical evidence, patients should understand that the evidence base for male use is limited to Phase 1/2 studies and clinical experience rather than the Phase 3 registration trials that supported the female HSDD approval. Discuss risks and benefits with a qualified healthcare provider before initiating therapy.
Male Sexual Health and the Broader Peptide Approach
For men interested in comprehensive sexual health optimization, bremelanotide is often discussed alongside other peptide therapies that target different aspects of male sexual and reproductive function. Understanding how these peptides relate to each other can help clinicians and patients develop integrated treatment strategies.
Gonadorelin, a synthetic form of gonadotropin-releasing hormone (GnRH), is used to stimulate the body's own production of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), which in turn support testosterone production and spermatogenesis. For men whose low desire is partly attributable to suboptimal testosterone levels, gonadorelin can address the hormonal component while bremelanotide addresses the dopaminergic component. This dual approach targets two distinct pathways influencing sexual desire.
Kisspeptin-10, another hypothalamic peptide, sits upstream of the GnRH system. Kisspeptin neurons in the hypothalamus integrate metabolic and reproductive signals, and kisspeptin administration has been shown to stimulate both hormone release and sexual arousal in human studies. The interaction between kisspeptin and melanocortin systems is an active area of investigation, with evidence suggesting that these pathways may cooperate in the central regulation of reproductive behavior.
Oxytocin, often called the "bonding hormone," plays complementary roles in sexual function. While bremelanotide primarily enhances desire and motivation, oxytocin facilitates the emotional connection, trust, and intimacy components of sexual experience. Some clinicians use oxytocin (nasal spray) in combination with bremelanotide to address both the motivational and emotional dimensions of sexual dysfunction. The melanocortin system has known interactions with oxytocin pathways in the hypothalamus, and these two peptide systems may have additive effects on sexual experience.
Growth hormone secretagogues like CJC-1295/Ipamorelin and MK-677 are sometimes used in the context of overall vitality and anti-aging protocols that indirectly support sexual function. Growth hormone supports lean body mass, energy levels, and tissue repair - all factors that can influence sexual health and self-confidence. While these peptides do not directly target sexual desire pathways, their effects on overall physical well-being can create a foundation that supports healthy sexual function.
For men dealing with the metabolic components that often accompany sexual dysfunction, GLP-1 receptor agonists like semaglutide and tirzepatide represent another relevant therapeutic category. Obesity is a significant risk factor for both erectile dysfunction and low testosterone, and weight loss through GLP-1 agonist therapy has been shown to improve both testosterone levels and sexual function. The GLP-1 weight loss overview provides additional information on how metabolic optimization can support sexual health outcomes.
Comparison to PDE5 Inhibitors
One of the most common questions from both clinicians and patients concerns how bremelanotide compares to PDE5 inhibitors - the dominant pharmacological class for sexual dysfunction treatment since sildenafil's approval in 1998. The comparison is instructive because it highlights fundamental differences in mechanism, indication, and clinical application that determine which patients will benefit from which approach.
Mechanism of Action: Central vs. Peripheral
The foundational difference between bremelanotide and PDE5 inhibitors lies in where they act within the sexual response pathway. PDE5 inhibitors work peripherally: they block the enzyme phosphodiesterase type 5 in the smooth muscle of the corpus cavernosum (in men) or clitoral and vaginal tissue (in women). By inhibiting PDE5, these drugs prevent the breakdown of cyclic guanosine monophosphate (cGMP), the intracellular messenger produced in response to nitric oxide release during sexual arousal. The result is enhanced vasodilation and blood engorgement of erectile tissue.
This peripheral mechanism means PDE5 inhibitors require pre-existing sexual arousal to work. They amplify the vascular response to arousal signals but do not create arousal or desire themselves. A man who takes sildenafil but feels no sexual desire will not achieve an erection. The drug only facilitates the physical response once the neural signal for arousal has been generated.
Bremelanotide, by contrast, works centrally. It activates MC4 receptors in the hypothalamus to stimulate dopamine release into motivation and reward circuits. This central action can generate desire and arousal independent of peripheral vascular factors. The drug works upstream of the point where PDE5 inhibitors act, addressing the motivational and psychological component of the sexual response rather than the vascular endpoint.
Head-to-Head Comparison Table
| Feature | Bremelanotide (PT-141) | Sildenafil (Viagra) | Tadalafil (Cialis) |
|---|---|---|---|
| Mechanism | MC4R agonist (central) | PDE5 inhibitor (peripheral) | PDE5 inhibitor (peripheral) |
| Primary target | Sexual desire and arousal | Erectile function | Erectile function |
| Route | Subcutaneous injection | Oral | Oral |
| Onset | 30-60 minutes | 30-60 minutes | 30-60 min (on-demand); continuous (daily) |
| Duration | ~6-8 hours | ~4-6 hours | ~24-36 hours (on-demand); continuous (daily) |
| Requires arousal | No - can generate desire | Yes - facilitates response to arousal | Yes - facilitates response to arousal |
| FDA-approved for | HSDD in premenopausal women | ED in men; PAH | ED in men; BPH; PAH |
| Efficacy in women | Demonstrated (Phase 3) | Not proven effective | Not proven effective |
| Food interactions | None | High-fat meals reduce absorption | Minimal |
| Alcohol interaction | No pharmacokinetic interaction | May worsen hypotension | May worsen hypotension |
| Nitrate contraindication | No | Yes - severe hypotension risk | Yes - severe hypotension risk |
| Main side effects | Nausea, flushing, headache | Headache, flushing, dyspepsia, visual changes | Headache, back pain, myalgia, nasal congestion |
| BP effect | Transient small increase | Decrease (especially with nitrates) | Decrease (especially with nitrates) |
| Max frequency | 8 doses/month (label) | Once daily | Once daily |
Why PDE5 Inhibitors Fail in Some Patients
Understanding why 30-40% of men fail PDE5 inhibitor therapy is essential for appreciating bremelanotide's potential role. PDE5 inhibitor failure can result from several factors, and the cause of failure determines whether a central-acting agent like bremelanotide might help.
Severe vascular disease: Men with advanced atherosclerosis, diabetic vasculopathy, or post-surgical vascular damage may have insufficient arterial inflow to generate an erection even with maximal cGMP enhancement. In these cases, bremelanotide alone may not solve the problem because the central arousal signal cannot overcome the peripheral vascular limitation. However, combination therapy (bremelanotide plus a PDE5 inhibitor) may partially restore function by maximizing both the central drive and the peripheral vascular response.
Neurogenic causes: Men who have undergone pelvic surgery (prostatectomy, cystectomy) or who have neurological conditions affecting the sacral nerve pathways may fail PDE5 inhibitors because the neural signals needed to trigger nitric oxide release are disrupted. Bremelanotide can generate central arousal signals, but if the peripheral neural pathway is interrupted, these signals may not fully translate to erectile function. Again, combination approaches may offer partial benefit.
Psychogenic causes: Men with performance anxiety, depression, or other psychological factors may fail PDE5 inhibitors because the drug cannot compensate for the central inhibition of arousal. This is where bremelanotide has the clearest theoretical advantage - by directly boosting dopaminergic motivation and reducing the threshold for arousal, it can overcome the psychological barriers that prevent the central arousal signal from being generated in the first place.
Low desire: Some men who present with erectile complaints actually have underlying low desire as the primary issue. They report "I can't get an erection" when the deeper problem is "I don't feel interested in sex." PDE5 inhibitors will not help these patients because the problem is not vascular insufficiency but rather absent motivational drive. Bremelanotide directly addresses this central desire deficit.
Female Sexual Dysfunction: Where PDE5 Inhibitors Fall Short
The comparison between bremelanotide and PDE5 inhibitors is particularly stark when considering female sexual dysfunction. Multiple clinical trials of sildenafil and other PDE5 inhibitors in women have produced disappointing results. While these drugs can increase genital blood flow and vaginal lubrication in women (as measured by vaginal photoplethysmography), they have consistently failed to improve subjective measures of desire, arousal, and satisfaction.
This disconnect between objective genital response and subjective sexual experience in women, sometimes called the arousal concordance gap, illustrates a fundamental principle: female sexual desire is predominantly a central nervous system phenomenon, not a peripheral vascular one. Increasing blood flow to the genitals does not make a woman feel desire. This is precisely why bremelanotide, which targets the central desire circuit, succeeds where PDE5 inhibitors fail in women.
For women with HSDD, bremelanotide is not just a better option than PDE5 inhibitors - it is essentially the only pharmacological approach that directly addresses the core pathophysiology. Flibanserin (Addyi) also works centrally but through serotonergic modulation rather than melanocortin activation, and it requires daily dosing rather than on-demand use.
Drug Interaction Profiles: Safety Advantages
Bremelanotide has a specifically cleaner drug interaction profile than PDE5 inhibitors. The most critical safety concern with PDE5 inhibitors is the absolute contraindication with nitrate medications (nitroglycerin, isosorbide mononitrate/dinitrate). Combining a PDE5 inhibitor with a nitrate can cause severe, potentially life-threatening hypotension. This contraindication excludes a substantial number of men with coronary artery disease from PDE5 inhibitor use.
Bremelanotide has no nitrate contraindication because it does not affect the nitric oxide-cGMP pathway. Men who take nitroglycerin or long-acting nitrates for angina can theoretically use bremelanotide without the hypotension risk inherent in PDE5 inhibitor use. This represents a significant safety advantage for patients with cardiovascular disease who also have sexual dysfunction.
Similarly, PDE5 inhibitors should be used cautiously with alpha-adrenergic blockers (commonly prescribed for benign prostatic hyperplasia) due to additive hypotensive effects. Bremelanotide does not share this interaction.
However, bremelanotide does carry its own cardiovascular caveat: it can cause transient blood pressure increases. While these are typically small (approximately 6/3 mmHg), they represent the opposite direction of PDE5 inhibitor-related blood pressure changes. Bremelanotide should be used cautiously in patients with uncontrolled hypertension and is contraindicated in those with uncontrolled blood pressure or cardiovascular disease.
Alcohol does not affect bremelanotide's pharmacokinetics or efficacy, a practical advantage since sexual activity often occurs in social settings where alcohol may be consumed. PDE5 inhibitors, while not contraindicated with moderate alcohol, can have additive blood pressure-lowering effects, and heavy alcohol consumption can impair erectile function regardless of medication use.
Practical Decision Framework for Clinicians
When deciding between bremelanotide and PDE5 inhibitors (or considering them in combination), clinicians should assess where in the sexual response cascade the patient's dysfunction occurs:
Choose bremelanotide when:
- The primary complaint is low desire ("I'm not interested in sex")
- The patient has HSDD (especially premenopausal women)
- PDE5 inhibitors have failed and psychogenic factors are suspected
- The patient takes nitrate medications
- The dysfunction is primarily central/motivational rather than vascular
Choose a PDE5 inhibitor when:
- The primary complaint is erectile inability ("I want to but can't")
- The patient has documented vascular ED
- Convenience of oral dosing is a priority
- The patient has normal desire but physical performance limitation
Consider combination therapy when:
- The patient has mixed desire and erectile dysfunction
- PDE5 inhibitors produce partial but insufficient response
- Post-prostatectomy patients with both desire and erectile issues
- SSRI-induced sexual dysfunction affecting both desire and function
The FormBlends dosing calculator can assist with individualized dosing for patients considering bremelanotide, while the science and research page provides additional mechanistic background.
The Complementary Approach
Rather than viewing bremelanotide and PDE5 inhibitors as competitors, the most nuanced clinical perspective treats them as complementary tools addressing different components of the sexual response. Just as a musician needs both the desire to play (motivation) and a functioning instrument (capability), sexual function requires both central desire/arousal signals and peripheral vascular response. Bremelanotide supports the former; PDE5 inhibitors support the latter. When both components are impaired, combination therapy may offer the best outcomes.
Clinical Case Scenarios
To illustrate the practical decision-making between bremelanotide and PDE5 inhibitors, consider the following representative clinical scenarios:
Scenario 1: Premenopausal Woman with Low Desire
A 38-year-old woman in a stable 12-year marriage reports that she has lost almost all interest in sexual activity over the past 2 years. She finds this distressing because she previously had a healthy sexual desire and her relationship is otherwise strong. Physical examination and laboratory work are normal. She meets criteria for acquired, generalized HSDD. Treatment: Bremelanotide 1.75 mg SC injection as needed. PDE5 inhibitors would not be appropriate because her problem is desire-based, not arousal or lubrication-based. She should be counseled about nausea management and the 45-minute advance dosing recommendation.
Scenario 2: Man with Vascular ED
A 62-year-old man with well-controlled type 2 diabetes and mild peripheral vascular disease reports progressive difficulty achieving and maintaining erections over 3 years. His desire for sex remains strong. He says, "I want to, but my body won't cooperate." Treatment: First-line PDE5 inhibitor (tadalafil 10-20 mg or sildenafil 50-100 mg). Bremelanotide alone would be insufficient because the primary problem is peripheral vascular, not central desire. If PDE5 inhibitors provide only partial response, combination therapy adding bremelanotide could be considered.
Scenario 3: Man with Performance Anxiety
A 35-year-old man reports erectile difficulty that began after a divorce. He has normal morning erections and can achieve erection with self-stimulation but loses his erection during partnered sexual activity. He describes significant anxiety about sexual performance. Treatment: Bremelanotide may be a better first option than a PDE5 inhibitor because the primary problem is psychogenic - central anxiety is inhibiting the arousal signal. By boosting dopaminergic motivation and reducing the arousal threshold, bremelanotide may help him "get out of his head." A PDE5 inhibitor could serve as an adjunct if needed.
Scenario 4: Man on Nitrates
A 58-year-old man with stable angina managed with isosorbide mononitrate reports erectile dysfunction. PDE5 inhibitors are absolutely contraindicated due to the nitrate interaction. Treatment: Bremelanotide is one of the few pharmacological options available because it has no nitrate contraindication. It can be used safely in patients taking nitrates, addressing a significant unmet need in this population. Careful blood pressure monitoring is recommended given his cardiovascular history.
Scenario 5: SSRI-Induced Sexual Dysfunction
A 42-year-old woman on sertraline for generalized anxiety disorder reports that her SSRI has eliminated her sexual desire. She is otherwise doing well on the medication and does not want to change it. Treatment: Bremelanotide as needed can address the SSRI-related desire suppression without requiring medication changes. The dopaminergic mechanism of bremelanotide may counteract the serotonergic suppression of desire circuits caused by the SSRI. This represents an off-label use that requires informed consent and clinical judgment.
Dosing & Administration
Bremelanotide is administered as an on-demand subcutaneous injection, a delivery method that allows for precise dosing and predictable pharmacokinetics. The FDA-approved Vyleesi product comes in a pre-filled autoinjector pen, while compounded formulations are typically supplied as reconstitutable lyophilized powder or ready-to-inject solutions in multi-dose vials. This section covers the practical details of dosing, administration technique, timing, and special considerations for both the approved and compounded formulations.
FDA-Approved Dosing (Vyleesi)
The recommended dose of Vyleesi is 1.75 mg administered subcutaneously at least 45 minutes before anticipated sexual activity. Each pre-filled syringe contains 1.75 mg of bremelanotide (equivalent to 1.89 mg bremelanotide acetate) in 0.3 mL of solution. The autoinjector pen is designed for single use and self-administration.
Key dosing parameters from the FDA label:
- Dose: 1.75 mg per injection
- Route: Subcutaneous (abdomen or thigh)
- Timing: At least 45 minutes before anticipated sexual activity
- Maximum frequency: No more than 1 dose per 24 hours
- Monthly limit: No more than 8 doses per month
- No dose adjustment: Required for hepatic or renal impairment (mild to moderate)
The 45-minute advance timing corresponds to the drug's pharmacokinetic profile. Following subcutaneous injection, bremelanotide reaches peak plasma concentration (Cmax) in approximately 1 hour, with a range of 0.5 to 1.0 hours. Many patients report onset of subjective effects (warmth, flushing, increased desire) within 30 to 45 minutes, supporting the recommended pre-activity timing.
Pharmacokinetic Profile
Understanding bremelanotide's pharmacokinetics helps clinicians and patients optimize timing and frequency of use:
| Parameter | Value |
|---|---|
| Bioavailability (subcutaneous) | 100% |
| Tmax (time to peak) | ~1 hour (range 0.5-1.0 hr) |
| Cmax | 72.8 ng/mL |
| AUC | 276 hr*ng/mL |
| Protein binding | 21% |
| Half-life | 2.5 hours |
| Clearance | 6.5 +/- 1.0 L/h |
| Renal excretion | 64.8% |
| Fecal excretion | 22.8% |
| Metabolism | Multiple hydrolysis reactions |
The 2.5-hour half-life means that plasma concentrations decline relatively quickly, with the drug largely cleared within 6 to 10 hours. This rapid clearance supports the on-demand dosing model and minimizes concerns about drug accumulation with repeated use. The 100% subcutaneous bioavailability ensures that the administered dose is fully absorbed, providing consistent and predictable drug exposure with each injection.
Bremelanotide is a 7-amino-acid cyclic peptide, and its metabolism occurs through multiple hydrolysis reactions - essentially, the body breaks down the peptide into its constituent amino acid components. There are no active metabolites of clinical significance, and the drug is not metabolized through cytochrome P450 enzymes. This means bremelanotide has very few drug-drug interactions based on metabolic competition, a practical advantage for patients taking multiple medications.
Injection Technique
For the Vyleesi autoinjector, the administration procedure is straightforward:
- Site selection: Choose an injection site on the abdomen (at least 2 inches from the navel) or the front of the thigh. Rotate injection sites with each use to minimize local reactions.
- Preparation: Remove the autoinjector from its packaging. Allow it to reach room temperature if refrigerated (approximately 30 minutes). Do not shake.
- Cleaning: Clean the injection site with an alcohol swab and allow it to dry completely.
- Injection: Remove the cap from the autoinjector. Place the device firmly against the skin at a 90-degree angle. Press the activation button. You will hear a click and feel a brief pinch as the needle deploys and the medication is injected.
- Post-injection: Hold the autoinjector in place for 5 seconds to ensure complete delivery. Remove and dispose of the autoinjector in a sharps container. Do not recap.
For compounded bremelanotide supplied in multi-dose vials, the injection technique uses standard subcutaneous injection practice with an insulin syringe (typically 29-31 gauge, 0.5 inch needle). Patients using compounded formulations should receive training on proper reconstitution (if using lyophilized powder), dose measurement, and injection technique from their prescribing clinician or pharmacist.
Compounded Formulation Dosing
Compounding pharmacies that supply PT-141 typically provide the peptide in several formulations, each with specific dosing considerations:
Subcutaneous Injection (reconstituted vial): The most common compounded formulation. Lyophilized bremelanotide powder is reconstituted with bacteriostatic water. Typical concentrations are 5 mg/mL or 10 mg/mL. The recommended starting dose for women is 1 to 2 mg (matching the FDA-approved 1.75 mg). For men using off-label, starting doses typically range from 1 to 2 mg, with some clinicians titrating up to 2.5 mg based on response and tolerability. The reconstituted solution should be stored refrigerated (2-8 degrees C) and used within 28 days.
Nasal Spray: Some compounding pharmacies prepare bremelanotide as a nasal spray, echoing the earlier clinical development formulation. Typical nasal doses are higher than subcutaneous doses (5-10 mg) because nasal bioavailability is lower. The nasal route offers needle-free convenience but has less predictable absorption and was associated with more pronounced blood pressure effects in the original clinical development program.
Sublingual Troches: Sublingual lozenges represent another needle-free alternative. Doses typically range from 1.5 to 2.5 mg. The sublingual route provides reasonable absorption through the oral mucosa, though bioavailability data specific to this formulation are limited. Troches may be combined with other compounds (e.g., bremelanotide plus tadalafil in a combination troche for men).
Timing and Practical Guidance
The official recommendation to inject at least 45 minutes before anticipated sexual activity is based on the drug's pharmacokinetic profile. However, individual response timing can vary:
Early responders: Some patients report feeling effects within 15 to 30 minutes. These individuals may not need the full 45-minute lead time.
Typical responders: Most patients notice a subjective increase in warmth, flushing, and sexual interest within 30 to 60 minutes. The 45-minute recommendation captures the majority of this response curve.
Late responders: A minority of patients report that peak subjective effects occur 60 to 90 minutes after injection. These individuals may benefit from administering the dose slightly earlier.
Patients should be advised that the drug's effects can last 6 to 8 hours, providing a generous window for sexual activity without the pressure of a narrow efficacy window. Unlike some medications where the effect wears off abruptly, bremelanotide's effects taper gradually as plasma concentrations decline.
Food does not affect bremelanotide's pharmacokinetics when administered subcutaneously, so timing relative to meals is not a concern. Alcohol consumption does not alter the drug's absorption or efficacy, though excessive alcohol can independently impair sexual function and exacerbate nausea.
Dose Titration and Individualization
The FDA-approved dose of 1.75 mg is fixed - the Vyleesi autoinjector does not allow dose adjustment. However, when using compounded formulations, clinicians have the flexibility to individualize dosing:
Starting dose: Most clinicians begin with 1 to 1.5 mg for both women and men, particularly in patients who are concerned about nausea or are small in body size.
Titration up: If the initial dose is tolerated but produces inadequate efficacy, the dose can be increased to 1.75 to 2 mg. Some male patients may be titrated to 2 to 2.5 mg under close supervision.
Nausea management: For patients who experience significant nausea at the standard dose, starting with a lower dose (0.5 to 1 mg) and gradually titrating upward over several sessions can help the body develop tolerance to the nauseogenic effect. Pre-treatment with an antiemetic such as ondansetron (Zofran) 4 mg taken 30 minutes before the bremelanotide injection can significantly reduce nausea.
Maximum dose: Clinical data support doses up to 2.5 mg subcutaneously, though doses above 1.75 mg are not FDA-approved and should be used with caution. Higher doses increase the risk of nausea and blood pressure elevation without necessarily providing proportionally greater efficacy.
Storage and Handling
Vyleesi autoinjectors should be stored at room temperature (20-25 degrees C / 68-77 degrees F) in their original packaging. They should be protected from light and should not be frozen. The product has a shelf life of approximately 24 months from the date of manufacture.
Compounded bremelanotide formulations have different storage requirements depending on the formulation:
- Lyophilized powder (before reconstitution): Can be stored at room temperature or refrigerated. Shelf life varies by pharmacy but is typically 6-12 months.
- Reconstituted solution: Must be refrigerated (2-8 degrees C / 36-46 degrees F). Typically stable for 28 days after reconstitution.
- Nasal sprays: Usually require refrigeration after first use. Typically stable for 30-60 days.
- Sublingual troches: Store at room temperature in a cool, dry place. Shelf life varies by pharmacy formulation.
For patients using compounded formulations from FormBlends, specific storage instructions will accompany the product. The dosing calculator can help determine appropriate reconstitution volumes for multi-dose vials, and the free assessment can connect patients with clinicians experienced in peptide therapy.
Dosing Quick Reference
- Standard dose: 1.75 mg subcutaneous
- When to inject: At least 45 minutes before sexual activity
- Max daily: 1 dose per 24 hours
- Max monthly: 8 doses per month
- Peak effect: 1 hour post-injection
- Duration: 6-8 hours
- Food/alcohol: No interactions
Reconstitution Guide for Compounded Formulations
Many patients receive bremelanotide as lyophilized powder in multi-dose vials from compounding pharmacies. Proper reconstitution is essential for accurate dosing and medication stability. Here is a step-by-step guide:
Step 1: Gather supplies. You will need the vial of lyophilized bremelanotide powder, a vial of bacteriostatic water (BAC water), an insulin syringe (typically 1 mL with 100-unit markings), and alcohol prep pads.
Step 2: Determine reconstitution volume. The reconstitution volume determines the concentration of your solution. For a 10 mg vial, adding 2 mL of BAC water gives a concentration of 5 mg/mL (each 0.1 mL = 0.5 mg). Adding 1 mL gives 10 mg/mL (each 0.1 mL = 1 mg). Common reconstitution volumes:
| Vial Size | BAC Water Added | Concentration | Volume for 1.75 mg Dose |
|---|---|---|---|
| 5 mg | 1 mL | 5 mg/mL | 0.35 mL (35 units) |
| 5 mg | 2 mL | 2.5 mg/mL | 0.70 mL (70 units) |
| 10 mg | 1 mL | 10 mg/mL | 0.175 mL (17.5 units) |
| 10 mg | 2 mL | 5 mg/mL | 0.35 mL (35 units) |
Step 3: Reconstitute. Wipe both vial tops with alcohol pads. Draw the desired volume of BAC water into the syringe. Insert the needle through the rubber stopper of the bremelanotide vial at a slight angle. Inject the water slowly down the side of the vial - do NOT squirt directly onto the powder, as this can damage the peptide. Gently roll the vial between your palms to mix. Do NOT shake. The powder should dissolve within 1-2 minutes, producing a clear, colorless solution.
Step 4: Storage. Store the reconstituted solution in the refrigerator (2-8 degrees C). Label the vial with the reconstitution date and discard after 28 days. Do not freeze reconstituted bremelanotide.
Step 5: Drawing a dose. Wipe the vial top with an alcohol pad. Using a clean insulin syringe, draw the appropriate volume for your prescribed dose. Tap the syringe to move any air bubbles to the top and push them out. Inject subcutaneously into the abdomen or thigh, rotating sites with each use.
The FormBlends dosing calculator can help determine the correct reconstitution volume and injection volume for your specific vial size and prescribed dose.
Patient Counseling Points
Effective patient counseling can significantly improve treatment outcomes and satisfaction with bremelanotide therapy. Key points to cover during the initial consultation include:
Set realistic expectations: Bremelanotide is not an aphrodisiac that will produce overwhelming desire or instant arousal. It provides a moderate enhancement of sexual interest and motivation. Most patients describe it as "feeling more open to sexual activity" or "noticing desire that wasn't there before" rather than an irresistible sexual urge. Approximately one in three women will experience a clearly meaningful improvement.
Nausea will likely improve: Many patients are discouraged by nausea with their first few doses. Counsel them that this is expected, typically diminishes substantially by the 4th-5th use, and can be managed with antiemetics. Ask patients to commit to at least 4-5 uses before deciding whether the drug works for them.
Timing matters: Encourage patients to experiment with injection timing. Some find that injecting 30 minutes before anticipated activity works well, while others prefer 60-90 minutes. The window of effect is generous (6-8 hours), so there is no need to rush.
Communication with partner: Encourage open communication between the patient and their partner about HSDD and its treatment. Partners who understand the condition and the treatment process tend to be more supportive, reducing performance pressure and improving outcomes.
It's not about needing a drug: Address any stigma the patient may feel about using medication for sexual desire. Frame bremelanotide as addressing a neurochemical imbalance, similar to how an antidepressant addresses a serotonin imbalance or an insulin injection addresses a metabolic deficiency. HSDD has a biological basis, and pharmacological treatment is a legitimate medical intervention.
Lifestyle factors still matter: Bremelanotide works best when combined with attention to sleep quality, stress management, relationship communication, and overall health. The drug enhances the brain's desire signal, but that signal works best in a context of general well-being and relational connection.
Safety Profile

Bremelanotide has been evaluated for safety across multiple clinical trials enrolling thousands of patients, with exposure durations up to 18 months in open-label extension studies. The safety database supports a well-characterized adverse effect profile that is manageable for most patients. This section provides a detailed review of common and uncommon side effects, cardiovascular considerations, the hyperpigmentation concern, contraindications, and practical strategies for minimizing adverse effects.
Common Adverse Effects
The most frequently reported adverse effects in the RECONNECT trials and the broader clinical development program are listed below, with their approximate incidence rates based on pooled data:
| Adverse Effect | Bremelanotide | Placebo |
|---|---|---|
| Nausea | 40% | 1% |
| Flushing | 20% | 1% |
| Injection site reactions | 13% | 8% |
| Headache | 11% | 8% |
| Nasopharyngitis | 3% | 3% |
| Upper respiratory infection | 3% | 3% |
| Fatigue | 3% | 1% |
| Hot flush | 3% | less than 1% |
| Dizziness | 2% | 1% |
| Back pain | 2% | 1% |
Nausea: The Most Significant Tolerability Concern
Nausea is by far the most common adverse effect and the primary reason patients discontinue bremelanotide therapy. The 40% incidence rate in clinical trials requires careful contextualization:
First-dose effect: Nausea is most pronounced with the first injection and typically decreases substantially with subsequent doses. Many patients who experience moderate nausea with their first 2-3 injections find that it diminishes to mild or absent nausea by the 4th or 5th use. This tachyphylaxis (tolerance development) to the nauseogenic effect is an important counseling point for patients and clinicians.
Timing: When nausea occurs, it typically begins within 30 to 60 minutes after injection, coinciding with peak plasma concentrations. The duration is usually 2 hours or less, though some patients report nausea lasting up to 4 hours.
Severity: In the RECONNECT trials, the majority of nausea events were rated as mild to moderate. Severe nausea requiring medical intervention was uncommon. Approximately 7% of bremelanotide-treated patients discontinued the drug due to nausea.
Mitigation strategies:
- Start with a lower dose (1 mg) and titrate upward as tolerance develops
- Pre-treat with ondansetron (Zofran) 4 mg orally 30 minutes before the bremelanotide injection
- Take the injection on a non-empty stomach (light snack 1-2 hours before)
- Avoid lying flat immediately after injection; remain upright for at least 30 minutes
- Avoid concurrent alcohol consumption, which can worsen nausea
- Use ginger supplements or ginger tea as a mild antiemetic
- Allow 4-5 uses before judging tolerability, as nausea often diminishes with repeated exposure
Flushing and Warmth
Flushing occurs in approximately 20% of patients and is related to MC1R activation and peripheral vasodilation. It typically manifests as a sensation of warmth or visible reddening of the face, neck, and upper chest. The effect usually begins within 30 minutes of injection and resolves within 2 to 4 hours.
While flushing is not medically dangerous, some patients find it socially inconvenient. The sensation of warmth, however, is sometimes described positively by patients as part of the drug's arousal-enhancing effect. No specific treatment is needed for flushing, and it tends to diminish with repeated use.
Cardiovascular Effects
Bremelanotide produces small, transient increases in blood pressure. The average change across the clinical program was approximately +6 mmHg systolic and +3 mmHg diastolic. These elevations typically occur within 2 to 4 hours of injection and resolve within 12 hours.
While a 6/3 mmHg increase is clinically insignificant for most healthy individuals, it raises concerns for specific populations:
Patients with uncontrolled hypertension: Bremelanotide is contraindicated in patients with uncontrolled hypertension. Blood pressure should be adequately controlled before initiating therapy.
Patients at high cardiovascular risk: The drug should be used with caution in patients with known cardiovascular disease, history of stroke, or multiple cardiovascular risk factors. While the blood pressure effect is small and transient, even small increases can theoretically trigger events in high-risk individuals.
Heart rate: Bremelanotide can produce small increases in heart rate (approximately 2-3 bpm on average). This effect is also transient and clinically insignificant for most patients.
It bears emphasis that bremelanotide's cardiovascular effect profile is opposite to that of PDE5 inhibitors, which lower blood pressure. This difference means that patients who are poor candidates for PDE5 inhibitors due to hypotensive risk (e.g., those on nitrates) may actually be better candidates for bremelanotide, while patients with uncontrolled hypertension should avoid bremelanotide but may tolerate PDE5 inhibitors.
Hyperpigmentation
Given bremelanotide's origin as a derivative of a tanning peptide, the potential for skin darkening was closely monitored throughout clinical development. The MC1R activity responsible for melanogenesis is retained in bremelanotide, and at sufficient exposure levels, the drug can stimulate melanin production.
In the RECONNECT trials, focal hyperpigmentation was reported by approximately 1% of patients who used bremelanotide at the recommended frequency (up to 8 doses per month). When it occurred, hyperpigmentation typically manifested as small, darkened areas on the face, gingiva (gums), or breasts. Patients with darker skin tones appeared to be at higher risk.
However, the hyperpigmentation risk increases substantially with more frequent dosing. In earlier clinical studies that used daily dosing regimens, hyperpigmentation occurred in more than one-third of subjects. This finding was a key factor in establishing the 8-dose-per-month maximum on the FDA label - a frequency that limits cumulative melanocortin stimulation and keeps hyperpigmentation risk low.
Hyperpigmentation associated with bremelanotide is generally reversible upon discontinuation, though resolution may take weeks to months. Patients should be counseled to report any new areas of skin darkening to their healthcare provider. The effect is cosmetic rather than medically harmful, but it can be distressing, particularly when it affects visible areas such as the face.
For patients interested in the tanning-related melanocortin compounds, the Melanotan II product page provides information about the parent compound with its stronger melanogenic activity.
Injection Site Reactions
Local injection site reactions were reported in approximately 13% of bremelanotide-treated patients versus 8% on placebo. These reactions typically included mild bruising, redness, itching, or discomfort at the injection site. They were generally mild, self-limiting, and rarely led to treatment discontinuation.
Proper injection technique, including rotation of injection sites, using appropriate needle gauge, and ensuring the alcohol prep pad is fully dry before injecting, can minimize local reactions. Patients who experience persistent injection site issues may benefit from applying ice to the area for 2 minutes before injection or using a topical numbing agent.
Contraindications and Precautions
Contraindications (absolute):
- Uncontrolled hypertension or known cardiovascular disease
- Hypersensitivity to bremelanotide or any component of the formulation
Precautions (use with caution):
- Patients at risk for cardiovascular disease
- Patients with hepatic or renal impairment (severe)
- Patients susceptible to hyperpigmentation (darker skin types)
- Patients taking naltrexone (bremelanotide may reduce naltrexone efficacy via opioid pathway interactions)
Drug interactions:
- Naltrexone: The FDA label includes a warning that bremelanotide may reduce the efficacy of naltrexone. Both drugs interact with endogenous opioid pathways, and concurrent use should be avoided. This is clinically relevant because some patients with sexual dysfunction may also be using naltrexone for alcohol use disorder or opioid use disorder.
- Other medications: No clinically significant interactions with CYP450-metabolized drugs, oral contraceptives, or common medications have been identified. Alcohol does not affect bremelanotide's pharmacokinetics.
Pregnancy and Lactation
Bremelanotide is not recommended during pregnancy. Animal reproductive studies showed decreased fetal survival at doses substantially higher than the human dose, though no structural malformations were observed. There are no adequate human data on bremelanotide use during pregnancy.
It is not known whether bremelanotide is excreted in human breast milk. Given the drug's peptide nature and its metabolism via hydrolysis, it is likely to be degraded in the gastrointestinal tract if ingested by a nursing infant. However, the absence of specific data means that caution is advised. The decision to breastfeed during bremelanotide therapy should consider the benefit of treatment to the mother against potential risks to the infant.
Long-Term Safety Data
The open-label extension study provided safety data for up to 18 months of bremelanotide exposure. Over this extended period:
- No new safety signals emerged compared to the 24-week blinded study period
- The incidence of nausea declined with continued use, supporting the tachyphylaxis effect
- Blood pressure effects remained small and transient with no evidence of cumulative cardiovascular harm
- Hyperpigmentation rates remained low at the recommended dosing frequency
- No hepatotoxicity, nephrotoxicity, or other organ toxicity was observed
- No impact on hormonal parameters (estrogen, progesterone, FSH, LH) was detected
A comprehensive safety analysis published by Clayton et al. in the Journal of Women's Health (2022) reviewed safety data across the entire clinical development program and concluded that bremelanotide was well-tolerated with a safety profile consistent across age groups, racial groups, and durations of exposure.
For patients using compounded formulations or considering bremelanotide for off-label indications, the same safety considerations apply. Regular monitoring of blood pressure is recommended, particularly during the first few doses. Patients with pre-existing cardiovascular risk factors should undergo a thorough evaluation before starting therapy. The free assessment at FormBlends can help connect patients with clinicians experienced in managing peptide therapies, while the science page provides additional safety and pharmacology information.
Safety Summary
Bremelanotide is generally well-tolerated, with nausea being the most common side effect (40%, improving with repeated use). Blood pressure increases are small and transient (+6/3 mmHg). Hyperpigmentation risk is low at recommended dosing frequencies (up to 8 doses/month). The drug is contraindicated with uncontrolled hypertension and should not be used concurrently with naltrexone. No serious organ toxicity has been observed in studies up to 18 months.
Special Populations & Clinical Considerations
Sexual dysfunction doesn't exist in a vacuum. It intersects with mental health, hormonal status, relationship dynamics, medications, and chronic illness in ways that make every person's experience unique. Bremelanotide's central mechanism of action, working through brain melanocortin pathways rather than peripheral vasculature, gives it potential applications across a wider range of clinical scenarios than PDE5 inhibitors. But it also means its effectiveness can be profoundly influenced by neurochemical context.
Antidepressant-Induced Sexual Dysfunction
Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) are among the most commonly prescribed medications worldwide, and sexual side effects affect 40-70% of users depending on the specific drug and how sexual dysfunction is measured. These effects include decreased libido, difficulty with arousal, delayed orgasm, and anorgasmia. For many patients, the sexual side effects are severe enough to cause medication non-adherence, which then leads to psychiatric relapse. It's a genuine clinical dilemma.
Bremelanotide is particularly relevant for this population because the mechanism of SSRI-induced sexual dysfunction involves serotonergic suppression of dopaminergic pathways in areas governing sexual motivation and reward. PT-141's activation of MC4R receptors promotes dopamine release in precisely these suppressed pathways. In theory, it bypasses the serotonergic blockade by activating an independent upstream pathway that feeds into the same dopaminergic circuits.
Clinical data specifically addressing bremelanotide in SSRI-treated patients are limited but encouraging. In the RECONNECT trials, participants were not excluded for SSRI use, and subgroup analyses suggested benefit in women taking antidepressants, though the subgroups weren't powered for definitive conclusions. Anecdotal reports from sexual medicine clinicians describe consistent benefit in SSRI-treated patients of both sexes, sometimes more pronounced than in non-medicated individuals, possibly because there's a clearer identifiable deficit to address.
For individuals on SSRIs experiencing sexual dysfunction, the practical approach involves using PT-141 at the standard 1.75 mg subcutaneous dose, administered 45-60 minutes before anticipated sexual activity. It's important to note that PT-141 won't counteract all SSRI-related sexual effects. While it can improve desire and arousal, difficulty reaching orgasm (a function more dependent on peripheral serotonergic effects) may persist. Setting realistic expectations matters. The goal is improvement, not complete reversal, of medication-induced sexual dysfunction.
The timing of PT-141 relative to SSRI dosing may also influence results. SSRIs produce varying levels of serotonergic suppression throughout the dosing interval, with peak suppression occurring 4-8 hours after oral administration (depending on the specific SSRI's pharmacokinetics). Scheduling sexual activity and PT-141 use during the trough of SSRI blood levels, typically in the morning for individuals who take their SSRI at bedtime, or in the evening for those who take their SSRI in the morning, may provide a slightly more favorable neurochemical context. This timing strategy is pragmatic rather than proven in clinical trials, but it aligns with basic pharmacokinetic principles.
Bupropion (Wellbutrin), a norepinephrine-dopamine reuptake inhibitor without serotonergic activity, is sometimes added to SSRI regimens specifically to counteract sexual side effects. The combination of bupropion plus on-demand PT-141 provides both continuous dopaminergic support (bupropion) and acute melanocortin-driven desire enhancement (PT-141) in the context of ongoing serotonergic treatment. This three-drug approach requires coordination with the prescribing psychiatrist but represents a sophisticated strategy for maintaining both mental health treatment and sexual function.
Postmenopausal Women
The Vyleesi FDA approval was specifically for premenopausal women with HSDD, leaving postmenopausal women technically outside the labeled indication. This was a regulatory decision based on the trial populations studied, not a pharmacological reason to expect the drug wouldn't work in postmenopausal women. MC4R receptor density in the brain doesn't decline with menopause in the same way that estrogen receptor signaling changes, which means the central mechanism should remain functional regardless of ovarian status.
Several factors make postmenopausal sexual dysfunction more complex than premenopausal HSDD. Declining estrogen causes vaginal atrophy, reduced lubrication, and sometimes pain with intercourse (dyspareunia). These are peripheral, tissue-level changes that PT-141's central mechanism doesn't directly address. A postmenopausal woman using bremelanotide might experience increased desire and mental arousal without corresponding physical readiness, which could be frustrating or even painful if peripheral tissue changes aren't also managed.
The optimal approach for postmenopausal women combines PT-141 for central desire enhancement with local estrogen therapy or other vaginal health interventions for peripheral tissue support. Vaginal DHEA (prasterone), low-dose vaginal estrogen, and hyaluronic acid-based moisturizers can maintain tissue health independently of systemic hormone levels. Oxytocin, which has documented effects on both bonding and genital tissue blood flow, may provide complementary benefit for postmenopausal women seeking a multi-faceted approach to sexual function restoration.
Testosterone replacement, either systemic at low doses or topical, is another consideration for postmenopausal women with low desire. Testosterone levels decline gradually throughout adulthood in women (they don't drop sharply at menopause like estrogen), and very low testosterone is associated with reduced sexual motivation independent of estrogen status. The combination of testosterone optimization plus on-demand PT-141 for specific occasions addresses both baseline hormonal support and acute desire enhancement. Hormone levels should be monitored by an experienced clinician. Visit the Peptide Research Hub for more on how peptide therapies complement hormonal approaches.
Men with Psychological Erectile Dysfunction
Erectile dysfunction in younger men (under 40) is predominantly psychological rather than vascular in origin. Performance anxiety, stress, depression, and relationship conflict account for the majority of ED cases in this demographic. PDE5 inhibitors like sildenafil work well for the physical mechanics but do nothing for the psychological component. A man might achieve an erection with Viagra but still feel no desire, creating a mechanical and emotionally unsatisfying experience.
PT-141 addresses this gap by working on the desire and motivation circuits. For men whose ED is rooted in performance anxiety, the experience of feeling genuine desire and mental arousal can break the anxiety cycle more effectively than achieving a pharmacologically-assisted erection. The brain realizes that desire is possible, the anxiety diminishes, and subsequent encounters become easier, sometimes to the point where ongoing pharmacological support isn't needed. Several sexual medicine specialists describe PT-141 as particularly valuable for "rebooting" sexual confidence in psychogenic ED cases.
The neuroplasticity component is worth emphasizing here. Psychogenic ED typically develops through a series of negative sexual experiences that strengthen inhibitory neural pathways. Each failed encounter reinforces the brain's association between sexual situations and anxiety or failure. PT-141 can help reverse this pattern by providing multiple positive experiences that strengthen facilitative neural pathways instead. Over time, these positive experiences can rewire the brain's default response to sexual situations from anxiety to anticipation, potentially creating lasting benefit that persists after the drug is discontinued.
The practical protocol for psychological ED typically involves PT-141 at 1-2 mg subcutaneously, taken 30-60 minutes before anticipated activity. Some clinicians combine a low dose of PT-141 (1 mg) with a low dose of a PDE5 inhibitor (sildenafil 25 mg) to address both the central desire component and any residual performance anxiety about physical response. This combination covers both pathways at sub-maximal doses of each, potentially reducing side effects from either drug compared to using higher doses of one alone.
Couples Dealing with Desire Discrepancy
One of the most common relationship complaints brought to sex therapists is mismatched desire levels between partners. When one partner consistently wants more sexual activity than the other, the resulting dynamic can create resentment, guilt, pressure, and avoidance that compounds the original desire gap. PT-141 offers a unique option for the lower-desire partner: pharmacological support for desire that doesn't feel like "going through the motions" because the compound generates genuine central arousal rather than simply enabling physical mechanics.
This application raises important ethical and relational questions that go beyond pharmacology. Using PT-141 should be a free choice by the lower-desire partner, not something imposed or expected by the higher-desire partner. The best outcomes occur when both partners understand the nature of the intervention, when it's part of a broader conversation about sexual health and relationship satisfaction, and when the lower-desire partner feels empowered rather than pressured by having this option available. Sexual medicine clinicians who prescribe PT-141 often recommend concurrent couples therapy to address the relational dynamics alongside the pharmacological intervention.
Individuals on GLP-1 Receptor Agonists
An increasingly relevant population includes individuals taking semaglutide, tirzepatide, or other GLP-1 receptor agonists for weight management. While these drugs effectively reduce body weight and improve metabolic health, some users report changes in libido during treatment. The mechanisms aren't fully understood but may involve hormonal changes from rapid fat loss (adipose tissue produces estrogen, and rapid fat loss can alter sex hormone levels), nausea affecting desire, or direct central effects of GLP-1 receptor activation on dopaminergic circuits.
Weight loss itself often improves sexual function through improved body image, better cardiovascular fitness, hormonal normalization (visceral fat produces excess estrogen through aromatase activity), and reduced inflammation. Many individuals find that as they lose weight on GLP-1 therapy, their baseline sexual function improves independent of any PT-141 use. But during the transition period, when the body is adapting to the medication and weight loss is actively occurring, PT-141 can bridge the gap between current sexual function and the improved function that metabolic health restoration will eventually provide.
For individuals experiencing decreased libido on GLP-1 therapy, PT-141 offers a targeted on-demand solution that works through an independent neurochemical pathway. There are no known pharmacological interactions between bremelanotide and GLP-1 receptor agonists, and the mechanisms of action don't overlap in ways that would suggest contraindication. The practical consideration is timing: if GLP-1-related nausea is present, adding PT-141 (which can also cause nausea) on the same day may compound this side effect. Scheduling PT-141 use on days when GLP-1-related nausea is minimal, typically 3-4 days after injection for weekly formulations, can minimize this overlap. Visit the GLP-1 Research Hub for more on managing side effects during GLP-1 therapy.
Practical Optimization & Troubleshooting
Bremelanotide is a powerful tool for sexual dysfunction, but getting the most out of it requires more than simply injecting the recommended dose and hoping for the best. Individual responses vary considerably, and understanding how to optimize your experience, manage side effects, and troubleshoot common problems makes the difference between a positive experience and a disappointing one.
Managing Nausea: The Primary Barrier to Satisfaction
Nausea is the most commonly cited reason for discontinuing PT-141, affecting roughly 40% of users in clinical trials. But with appropriate strategies, most people can reduce nausea to a manageable level or eliminate it entirely. Understanding why it happens helps frame the solutions.
PT-141 activates melanocortin receptors in the area postrema, a brain region involved in nausea and vomiting that lies outside the blood-brain barrier. This is the same reason other melanocortin-active peptides like Melanotan II also cause nausea. The good news is that this response typically diminishes with repeated exposures as the area postrema habituates to MC4R activation.
Dose titration: Starting at a lower dose (0.5-1.0 mg instead of the full 1.75 mg) for the first two or three uses allows the nausea response to attenuate before reaching the therapeutically optimal dose. Many clinicians now recommend this gradual approach as standard practice rather than starting at the full dose.
Timing relative to food: Taking PT-141 on a completely empty stomach worsens nausea for most people. A light meal 1-2 hours before administration, avoiding heavy, fatty, or spicy foods, provides some protection. Having ginger tea or ginger chews available is a practical strategy, as ginger has well-documented antiemetic properties that work through a different mechanism than pharmaceutical antiemetics.
Ondansetron (Zofran) pre-treatment: For individuals who experience significant nausea despite dose titration and dietary timing, taking ondansetron 4 mg orally 30 minutes before PT-141 injection effectively prevents nausea in most cases. This is the same antiemetic commonly used in chemotherapy and post-surgical settings. Some compounding pharmacies offer PT-141 combined with ondansetron for convenience. Discuss this option with your prescriber if nausea remains problematic.
Injection site considerations: Some users report that injecting into the thigh causes more nausea than abdominal injection, possibly due to differences in absorption rate. Abdominal subcutaneous injection provides slower, more gradual absorption, which may produce a less acute spike in MC4R activation and correspondingly less nausea. Rotating injection sites within the abdominal area (avoiding the two-inch radius around the navel) is the most commonly recommended approach.
Optimizing Timing and Setting
The timing of PT-141 administration relative to sexual activity significantly affects the experience. Clinical trials instructed participants to self-administer at least 45 minutes before anticipated activity. But individual pharmacokinetics vary, and some people find their optimal window is different from the average.
Onset timing: Most users notice the first effects (increased mental focus on sexual thoughts, heightened awareness of touch, genital warmth or tingling) between 30 and 90 minutes after injection. The peak effect typically occurs 1-2 hours post-injection and can persist for 6-12 hours. Planning accordingly means administration should be timed to the beginning of an evening together, not immediately before getting into bed.
Setting matters more than with PDE5 inhibitors: Because PT-141 works on desire and motivation centers in the brain, the psychological and environmental context significantly influences its effectiveness. Taking PT-141 in a relaxed, low-stress setting where emotional connection is possible produces better results than using it when rushed, anxious, or distracted. This isn't a failing of the drug; it's a reflection of how central desire pathways work. They respond to context. A warm bath, unhurried conversation, gentle physical contact, or whatever creates a sense of intimacy for a particular couple all complement PT-141's neurochemical effects.
Frequency considerations: The FDA labeling for Vyleesi recommends no more than one dose per 24 hours and no more than 8 doses per month. The 24-hour restriction relates to cardiovascular safety (specifically, avoiding cumulative blood pressure effects). The monthly limitation is primarily about minimizing hyperpigmentation risk with repeated melanocortin receptor activation. For most users, one to four doses per month provides the on-demand support they need. Using it at every sexual encounter is neither necessary nor recommended, as many couples find that the confidence gained from successful PT-141-assisted encounters carries over into unassisted ones.
When PT-141 Doesn't Work: Diagnostic Considerations
About 25-30% of women in the RECONNECT trials didn't show clinically meaningful improvement with bremelanotide. Non-response doesn't mean the drug "doesn't work." It usually means the primary driver of sexual dysfunction lies outside the MC4R-dopaminergic pathway that PT-141 targets. Understanding why it isn't working points toward more appropriate interventions.
Pain-dominant dysfunction: If the primary barrier to sexual function is pain rather than desire (dyspareunia, vaginismus, vulvodynia), PT-141 may increase desire without addressing the physical barrier, creating a frustrating disconnect. Pain conditions require their own targeted treatment: pelvic floor physical therapy, local anesthetics, estrogen or DHEA for atrophic changes, or specialized pain management approaches.
Relationship distress: When the core issue is relational rather than neurochemical, no medication can substitute for addressing communication failures, trust violations, or unresolved conflict. Individuals who feel no desire specifically toward their current partner but experience normal desire in other contexts (fantasy, new attraction) are dealing with a relational dynamic, not a neurochemical deficit. Couples therapy, not pharmacotherapy, is the appropriate first-line intervention.
Severe hormonal deficiency: Very low testosterone in either sex, or severely depleted estrogen in women, creates a hormonal environment that may not respond adequately to MC4R stimulation. If baseline hormone levels haven't been checked, this is a necessary step before concluding that PT-141 isn't effective. Optimizing the hormonal foundation often makes PT-141 more effective when subsequently tried.
Neurological conditions: Conditions affecting the hypothalamus, dopaminergic pathways, or melanocortin system (including certain pituitary disorders, Parkinson's disease, and some traumatic brain injuries) may impair the neural circuitry that PT-141 depends on. These situations require specialist evaluation.
Combining PT-141 with Other Approaches
Sexual function is multi-dimensional, and the most effective approaches often combine targeted pharmacology with behavioral, relational, and lifestyle interventions.
PT-141 plus testosterone optimization: For both men and women with low testosterone, optimizing testosterone levels provides a hormonal foundation that enhances PT-141's acute effects. Think of testosterone as setting the "baseline" level of sexual motivation, while PT-141 provides an acute boost above that baseline. With a higher baseline, the same PT-141 dose produces a more pronounced effect. Gonadorelin can support endogenous testosterone production in men, offering a way to optimize hormonal status without suppressing natural production.
PT-141 plus mindfulness-based sex therapy: Mindfulness techniques for sexual function, including sensate focus exercises and body awareness practices, enhance the subjective experience of PT-141's neurochemical effects. The drug increases the signal; mindfulness reduces the noise. Together, they produce a clearer, more integrated experience of desire and arousal than either approach alone.
PT-141 plus pelvic floor rehabilitation: For women with hypertonic pelvic floor (often from chronic stress, trauma, or habitual guarding against anticipated pain), pelvic floor physical therapy addresses the muscular tension that can prevent physical arousal even when mental desire is present. Combining PT-141 with a pelvic floor program addresses both the central motivation and the peripheral muscular barriers.
Sexual health is a component of overall wellbeing that intersects with sleep, stress management, hormonal balance, and physical fitness. For individuals pursuing comprehensive optimization, the FormBlends assessment can help identify which compounds and protocols best support your specific goals across these interconnected domains.
Emerging Research & Future Directions in Melanocortin Sexual Medicine
The development of bremelanotide was just the beginning of melanocortin-based sexual medicine. Ongoing research is exploring new delivery systems, next-generation compounds, and expanded indications that could dramatically broaden the therapeutic applications of MC4R-targeting agents.
Next-Generation Melanocortin Compounds
Several pharmaceutical companies and academic laboratories are developing melanocortin receptor agonists with improved selectivity, reduced side effects, and alternative delivery routes. The ideal next-generation compound would selectively activate MC4R in brain regions governing sexual function without activating MC4R in the area postrema (which causes nausea) or melanocytes (which causes hyperpigmentation).
Functionally selective (or "biased") MC4R agonists represent one approach. These compounds bind to the same receptor but activate different downstream signaling cascades depending on the tissue context. By designing molecules that preferentially activate Gq-protein signaling (associated with sexual arousal effects) over beta-arrestin signaling (associated with some side effects), researchers aim to separate the desired sexual effects from unwanted nausea and cardiovascular effects. This approach is still in early research stages, but the concept has been validated for other G-protein-coupled receptor targets.
Intranasal delivery of melanocortin agonists is another area of active investigation. Nasal administration bypasses first-pass metabolism, potentially allows more direct access to brain targets through the olfactory neural pathway, and could eliminate the need for injection. Early research on nasally administered bremelanotide suggests adequate absorption and brain penetration, though optimizing the formulation for consistent dosing and acceptable nasal tolerability requires additional development work. An intranasal product would dramatically lower the barrier to use, since nasal sprays are familiar and socially acceptable in a way that self-injection may not be for all patients.
Topical formulations applied to genital tissue represent yet another approach. While bremelanotide's primary mechanism is central (brain-mediated), local melanocortin receptors exist in genital tissue, and local application could provide both central effects (through transdermal absorption into systemic circulation) and direct peripheral effects (enhanced local blood flow and sensitivity). Topical formulations would be the most patient-friendly delivery option, potentially incorporated into intimate products that don't require any medical paraphernalia.
Expanded Indications Beyond HSDD
Research is exploring melanocortin receptor modulation for sexual dysfunction contexts beyond the approved HSDD indication. Several areas show particular promise:
Orgasmic dysfunction: Anorgasmia, the persistent inability to reach orgasm despite adequate stimulation and arousal, affects approximately 10-15% of women and a smaller but significant percentage of men. Current treatments are limited. Preliminary research suggests MC4R activation may facilitate the neurological pathways involved in orgasm, though the data are early and mechanism is not fully understood. Case reports from clinicians using bremelanotide describe improved orgasmic function in some patients, but controlled trials specifically targeting anorgasmia haven't been conducted.
Sexual dysfunction in cancer survivors: Cancer treatments, including chemotherapy, radiation, hormone therapy, and surgery, frequently cause sexual dysfunction through multiple mechanisms. The neurological, hormonal, and psychological impacts of cancer treatment create a complex sexual dysfunction picture that single-mechanism treatments often fail to address adequately. PT-141's central mechanism may provide benefit in this population by enhancing the desire and arousal components that hormonal deficits and psychological trauma impair, while being compatible with ongoing cancer monitoring and treatment. Oxytocin may complement PT-141 in cancer survivor sexual rehabilitation by addressing the intimacy and bonding aspects that cancer treatment can disrupt.
Sexual dysfunction in spinal cord injury: Individuals with incomplete spinal cord injury often retain some sexual function that can be enhanced pharmacologically. PT-141's central mechanism means it doesn't require intact genital neurological pathways for the desire and arousal components of its effect. While the physical sexual response may be limited by the injury, enhanced central arousal and desire can improve the subjective sexual experience and satisfaction. Research in this population is limited but conceptually supported by the mechanism of action.
The Psychology-Pharmacology Interface
Perhaps the most interesting emerging direction in PT-141 research isn't about new molecules or delivery systems but about how pharmacological intervention interacts with psychological processes over time. Clinicians who have used bremelanotide for years report a consistent pattern: some patients use the drug for a period, experience restored sexual confidence, and eventually reduce or discontinue use while maintaining improved sexual function. The drug appears to have helped "reset" a psychological pattern that was self-perpetuating.
This observation aligns with neuroscience research showing that repeated experiences of desire and arousal can strengthen the neural pathways supporting those experiences, a process called experience-dependent plasticity. If anxiety or negative associations have weakened sexual arousal pathways, pharmacologically induced positive sexual experiences may rebuild them. The drug provides the initial "push" that natural neuroplasticity then maintains.
This potential for lasting benefit beyond the period of active use distinguishes PT-141 from most pharmacological treatments for sexual dysfunction. PDE5 inhibitors work only while the drug is active; there's no lasting neuroplastic benefit from repeated sildenafil use. But a centrally acting agent that restores desire and arousal through brain pathways may produce durable changes in those pathways through normal learning and reinforcement processes. If validated by controlled research, this "pharmacological priming" concept could change how PT-141 is prescribed: not as ongoing symptomatic therapy but as a time-limited intervention designed to trigger lasting neurological recalibration.
Understanding the full potential of melanocortin-based sexual medicine requires continued research in all these directions. For individuals exploring PT-141 today, the FormBlends PT-141 product page provides current information on compound specifications and quality standards, while the Peptide Research Hub tracks emerging research across the peptide therapeutics space. The free assessment can help determine whether PT-141, alone or in combination with complementary approaches, is appropriate for your specific situation.
The Relationship Between Metabolic Health and Sexual Function
Sexual dysfunction and metabolic disease are deeply interconnected, yet they're typically treated by different specialists who may not coordinate care. Obesity impairs sexual function through multiple mechanisms: excess visceral fat converts testosterone to estrogen through aromatase activity, insulin resistance damages vascular endothelium needed for genital blood flow, inflammatory cytokines from adipose tissue suppress hypothalamic sexual motivation centers, and the psychological burden of weight-related body image concerns creates performance anxiety that further inhibits sexual response.
For individuals addressing metabolic health with GLP-1 receptor agonists while also managing sexual dysfunction with PT-141, the metabolic improvement often creates a positive trajectory for sexual function over time. As visceral fat decreases, testosterone levels typically rise in both men and women. As insulin sensitivity improves, vascular function recovers, supporting better genital blood flow. As systemic inflammation decreases, the neuroinflammatory suppression of sexual motivation pathways may resolve. Some patients find that after 6-12 months of successful metabolic treatment with semaglutide or tirzepatide, their need for PT-141 decreases as underlying metabolic contributors to sexual dysfunction are resolved.
This interconnection underscores the importance of viewing sexual health within the broader context of overall metabolic and hormonal health rather than as an isolated symptom requiring an isolated treatment. The GLP-1 Research Hub and the Lifestyle Hub provide resources on the metabolic and lifestyle foundations that support sexual health alongside pharmacological interventions.
Exercise, Fitness, and Sexual Function
Regular physical exercise is one of the most potent natural enhancers of sexual function, working through mechanisms that complement PT-141's pharmacological effects. Aerobic exercise improves cardiovascular fitness and vascular endothelial function, enhancing blood flow to genital tissue. Resistance training increases testosterone production in both men and women, supporting baseline libido. Exercise also promotes dopamine release in the brain's reward circuits, the same pathways that PT-141 activates through MC4R stimulation. And the psychological benefits of exercise, improved body image, reduced anxiety, enhanced self-efficacy, address the cognitive and emotional components of sexual dysfunction that pharmacology alone may not fully resolve.
For individuals using PT-141, maintaining a regular exercise program amplifies the drug's effects in several ways. Better cardiovascular fitness means better genital blood flow when PT-141-enhanced central arousal generates the demand for physical response. Higher baseline testosterone means a stronger hormonal foundation for PT-141's melanocortin activation to build upon. Improved body confidence reduces the performance anxiety that can interfere with sexual response. And the stress-reducing effects of regular exercise lower cortisol levels that otherwise suppress the hypothalamic sexual motivation circuits PT-141 targets.
The timing of exercise relative to PT-141 use may also matter. Some individuals report enhanced sexual responsiveness in the hours following vigorous exercise, possibly due to elevated dopamine, endorphins, and testosterone. If practical, scheduling exercise earlier in the day and PT-141 use for the evening may create an optimal neurochemical environment. Conversely, exhausting exercise immediately before sexual activity can leave individuals too physically depleted to benefit from PT-141's central arousal effects. Finding the balance between exercise-enhanced physiology and adequate physical energy for sexual activity is an individual optimization process.
Storage, Reconstitution, and Quality Considerations
For individuals using compounded PT-141 rather than the commercially available Vyleesi autoinjector, proper handling is essential for consistent results. PT-141 is supplied as a lyophilized powder that must be reconstituted with bacteriostatic water before use. The reconstituted solution should be refrigerated at 2-8 degrees Celsius and used within 28 days. Exposure to heat, light, or excessive agitation degrades the peptide, reducing potency and potentially altering its effects. Always use a new sterile needle for each injection, clean the vial stopper with an alcohol swab before each withdrawal, and discard any solution that appears cloudy or discolored. FormBlends provides pharmaceutical-grade PT-141 with documented purity testing, ensuring consistent compound quality across uses.
PT-141 and Relationship Dynamics: The Interpersonal Dimension
Sexual dysfunction rarely exists in a vacuum, and the decision to use PT-141 inevitably intersects with relationship dynamics, communication patterns, and the emotional landscape shared between partners. While the pharmacological mechanism of PT-141 is well-defined, its real-world impact depends heavily on the interpersonal context in which it's used. Addressing this dimension openly helps patients achieve better outcomes and avoid the disappointment that can follow when a medication is expected to solve problems that extend beyond biology.
One common pattern that clinicians observe is the "spectator effect" in long-term relationships where sexual dysfunction has been present for months or years. Both partners develop anxiety around sexual encounters, with the affected partner focusing intensely on whether the medication will "work" and the other partner monitoring for signs of arousal rather than relaxing into the experience. This mutual spectatoring creates performance pressure that can counteract PT-141's central arousal effects, since the melanocortin system responds to psychological context as well as pharmacological input. Couples who discuss PT-141 use openly, set expectations together, and agree to approach the experience with curiosity rather than pressure consistently report better outcomes than individuals who use the medication without their partner's knowledge or involvement.
For women using PT-141 for hypoactive sexual desire disorder (HSDD), the relationship dimension is especially relevant. Female sexual desire is influenced by a complex interplay of biological, psychological, and relational factors, and research consistently shows that relationship satisfaction is one of the strongest predictors of sexual desire in women. PT-141 addresses the neurochemical component of desire by activating melanocortin-4 receptors in the hypothalamus, but it cannot compensate for unresolved relationship conflicts, communication breakdowns, or emotional disconnection. Women who report the best outcomes with PT-141 typically describe using it within the context of a relationship where emotional intimacy is intact and where the medication supplements an existing foundation of connection rather than attempting to replace it.
The disclosure conversation itself, telling a partner about PT-141 use, can be surprisingly therapeutic. Many couples find that discussing sexual difficulties openly for the first time creates a sense of partnership around the issue that reduces shame and isolation. Partners who learn about PT-141's mechanism of action (central arousal rather than peripheral blood flow) often feel reassured that the medication enhances genuine desire rather than creating an artificial response. This understanding can transform the experience from "needing a drug to want me" into "investing in our connection by addressing a biological barrier," a reframing that benefits both partners psychologically.
Timing and communication around PT-141 use also matter practically. Because PT-141 requires 45-60 minutes to reach peak effect and its window of enhanced arousal can last 6-12 hours, couples benefit from discussing the general plan rather than treating each use as a surprise. Some couples develop simple signals or routines that indicate PT-141 has been administered, allowing the non-using partner to participate in creating conditions conducive to sexual activity, such as arranging privacy, reducing distractions, or engaging in the kind of emotional and physical affection that supports the transition from daily routine to intimate connection.
For same-sex couples, the dynamics around PT-141 use may differ in certain respects. Same-sex male couples dealing with erectile dysfunction or desire discrepancy often face the additional pressure of sexual performance expectations within gay male culture, where sexual vitality is heavily valued. PT-141's central arousal mechanism, which enhances desire and motivation rather than simply producing an erection, can be particularly well-suited for addressing the desire component that PDE5 inhibitors like sildenafil do not target. Same-sex female couples dealing with desire discrepancy may find PT-141 helpful for the lower-desire partner, though the research on PT-141 in this specific population is limited. In all cases, open communication about medication use and its intended effects supports better outcomes regardless of relationship structure. When relationship issues are significant enough that PT-141 alone is unlikely to produce satisfactory results, combination approaches that include couples therapy or sex therapy alongside pharmacological intervention tend to be most effective. A skilled sex therapist can help couples address the communication patterns, body image concerns, past sexual trauma, and desire discrepancies that commonly accompany sexual dysfunction. PT-141 serves as one tool within this broader therapeutic framework, providing the neurochemical support that makes it easier to engage with the psychological and relational work. The FormBlends assessment evaluates sexual health within the context of overall wellbeing, and the PT-141 product page provides practical guidance on administration and dosing for both men and women.
Frequently Asked Questions
References
- Kingsberg SA, Clayton AH, Pfaus JG. The female sexual response: current models, neurobiological underpinnings and agents currently approved or under investigation for the treatment of hypoactive sexual desire disorder. CNS Drugs. 2015;29(11):915-933. DOI: 10.1007/s40263-015-0288-1.
- Clayton AH, Althof SE, Kingsberg S, et al. Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo-controlled dose-finding trial. Women's Health. 2016;12(3):325-337. DOI: 10.2217/whe-2016-0018.
- Kingsberg SA, Clayton AH, Portman D, et al. Bremelanotide for the treatment of hypoactive sexual desire disorder: two randomized phase 3 trials. Obstetrics & Gynecology. 2019;134(5):899-908. DOI: 10.1097/AOG.0000000000003500. PMID: 31599840.
- Simon JA, Kingsberg SA, Portman D, et al. Long-term safety and efficacy of bremelanotide for hypoactive sexual desire disorder. Obstetrics & Gynecology. 2019;134(5):909-917. DOI: 10.1097/AOG.0000000000003514. PMID: 31599847.
- Diamond LE, Earle DC, Heiman JR, et al. An effect on the subjective sexual response in premenopausal women with sexual arousal disorder by bremelanotide (PT-141), a melanocortin receptor agonist. Journal of Sexual Medicine. 2006;3(4):628-638. DOI: 10.1111/j.1743-6109.2006.00268.x.
- Diamond LE, Earle DC, Rosen RC, et al. Double-blind, placebo-controlled evaluation of the safety, pharmacokinetic properties and pharmacodynamic effects of intranasal PT-141, a melanocortin receptor agonist, in healthy males and patients with mild-to-moderate erectile dysfunction. International Journal of Impotence Research. 2004;16(1):51-59. DOI: 10.1038/sj.ijir.3901139. PMID: 14963471.
- Safarinejad MR. Salvage of sildenafil failures with bremelanotide: a randomized, double-blind, placebo controlled study. Journal of Urology. 2008;179(3):1066-1071. DOI: 10.1016/j.juro.2007.10.063. PMID: 18206919.
- Pfaus JG, Shadiack A, Van Soest T, et al. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proceedings of the National Academy of Sciences. 2004;101(27):10201-10204. DOI: 10.1073/pnas.0400491101.
- Hadley ME. Discovery that a melanocortin regulates sexual functions in male and female humans. Peptides. 2005;26(10):1687-1689. DOI: 10.1016/j.peptides.2005.01.023.
- Molinoff PB, Shadiack AM, Earle D, et al. PT-141: a melanocortin agonist for the treatment of sexual dysfunction. Annals of the New York Academy of Sciences. 2003;994:96-102. DOI: 10.1111/j.1749-6632.2003.tb03167.x.
- Pfaus JG. Pathways of sexual desire. Journal of Sexual Medicine. 2009;6(6):1506-1533. DOI: 10.1111/j.1743-6109.2009.01309.x.
- Clayton AH, Kingsberg SA, Portman D, et al. Safety profile of bremelanotide across the clinical development program. Journal of Women's Health. 2022;31(2):171-182. DOI: 10.1089/jwh.2021.0191. PMID: 35147466.
- Thomas A, Woodard C, Rovner ES, et al. Urologic complications of nonurologic medications. Urologic Clinics of North America. 2003;30(1):123-131.
- Arnow BA, Millheiser L, Garrett A, et al. Women with hypoactive sexual desire disorder compared to normal females: a functional magnetic resonance imaging study. Neuroscience. 2009;158(2):484-502. DOI: 10.1016/j.neuroscience.2008.09.044.
- Mayer D, Lynch SE. Bremelanotide: new drug approved for treating hypoactive sexual desire disorder. Annals of Pharmacotherapy. 2020;54(7):684-690. DOI: 10.1177/1060028019899152.
- Portman DJ, Clayton AH, Engelen S, et al. Reliability and validity of the elements of desire questionnaire in premenopausal women with hypoactive sexual desire disorder. Journal of Sexual Medicine. 2020;17(11):2219-2228. DOI: 10.1016/j.jsxm.2020.07.070.
- Kingsberg SA, Woodard T. Female sexual dysfunction: focus on low desire. Obstetrics & Gynecology. 2015;125(2):477-486. DOI: 10.1097/AOG.0000000000000620.
- Uckert S, Bannowsky A, Albrecht K, et al. Melanocortin receptor agonists in the treatment of male and female sexual dysfunctions: results from basic research and clinical studies. Expert Opinion on Investigational Drugs. 2014;23(11):1477-1483. DOI: 10.1517/13543784.2014.934805.
- Rosen RC, Riley A, Wagner G, et al. The International Index of Erectile Function (IIEF): a multidimensional scale for assessment of erectile dysfunction. Urology. 1997;49(6):822-830. DOI: 10.1016/S0090-4295(97)00238-0.
- Hruby VJ. Designing peptide receptor agonists and antagonists. Nature Reviews Drug Discovery. 2002;1(11):847-858. DOI: 10.1038/nrd939.
- Wessells H, Fuciarelli K, Hansen J, et al. Synthetic melanotropic peptide initiates erections in men with psychogenic erectile dysfunction: double-blind, placebo controlled crossover study. Journal of Urology. 1998;160(2):389-393. PMID: 9679884.
- Goldstein I, Kim NN, Clayton AH, et al. Hypoactive sexual desire disorder: International Society for the Study of Women's Sexual Health (ISSWSH) expert consensus panel review. Mayo Clinic Proceedings. 2017;92(1):114-128. DOI: 10.1016/j.mayocp.2016.09.018.
- Perelman MA. The sexual tipping point: a mind/body model for sexual medicine. Journal of Sexual Medicine. 2009;6(3):629-632. DOI: 10.1111/j.1743-6109.2008.01177.x.
- Montague DK, Jarow JP, Broderick GA, et al. The management of erectile dysfunction: an AUA update. Journal of Urology. 2005;174(1):230-239. DOI: 10.1097/01.ju.0000164463.19239.19.
- Kingsberg SA, Althof SE, Simon JA, et al. Female sexual dysfunction - medical and psychological treatments, committee 14. Journal of Sexual Medicine. 2017;14(12):1463-1491. DOI: 10.1016/j.jsxm.2017.05.018.
- Dhillon S, Keam SJ. Bremelanotide: first approval. Drugs. 2019;79(14):1599-1606. DOI: 10.1007/s40265-019-01187-w. PMID: 31429064.
- Koochaki PE, Kingsberg SA, Simon JA, et al. Pathways linking menopause-related factors to hypoactive sexual desire disorder in postmenopausal women. Journal of Clinical Endocrinology & Metabolism. 2021;106(4):e1640-e1650.
- Palatin Technologies. Palatin announces publication of Vyleesi (bremelanotide) fMRI study in women with hypoactive sexual desire disorder (HSDD). Press Release, February 2022.
- Lee JY, Kim SW, Paick JS. Novel emerging therapies for erectile dysfunction. World Journal of Men's Health. 2021;39(1):44-64. DOI: 10.5534/wjmh.200007.
- Jaspers L, Feys F, Bramer WM, et al. Efficacy and safety of flibanserin for the treatment of hypoactive sexual desire disorder in women: a systematic review and meta-analysis. JAMA Internal Medicine. 2016;176(4):453-462. DOI: 10.1001/jamainternmed.2015.8565.
- DeRogatis LR, Komer L, Katz M, et al. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. Journal of Sexual Medicine. 2012;9(4):1074-1085.
- Jordan R, Krop J, Schaffer S, et al. The neurobiology of bremelanotide for the treatment of hypoactive sexual desire disorder in premenopausal women. CNS Spectrums. 2021;26(5):514-523. DOI: 10.1017/S1092852920002096. PMID: 33455598.
- Rosen RC, Brown C, Heiman J, et al. The Female Sexual Function Index (FSFI): a multidimensional self-report instrument for the assessment of female sexual function. Journal of Sex & Marital Therapy. 2000;26(2):191-208. DOI: 10.1080/009262300278597.
- Shifren JL, Monz BU, Russo PA, et al. Sexual problems and distress in United States women: prevalence and correlates. Obstetrics & Gynecology. 2008;112(5):970-978. DOI: 10.1097/AOG.0b013e3181898cdb.
- Katz M, DeRogatis LR, Ackerman R, et al. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. Journal of Sexual Medicine. 2013;10(7):1807-1815.